An Introduction to Protein Expression
For ordering information on the products discussed here, please visit our Protein Expression product pages.
Introduction to Translation Biology
Cell-free protein synthesis (CFPS), also known as in vitro transcription/translation, is an important and versatile staple in the collection of tools available to molecular biologists for the elucidation of cellular pathways and mechanisms in fundamental and applied sciences. It has become an increasingly popular and useful approach in both high-throughput functional genomics and proteomics, offering significant advantages over protein expression performed in live cells.
Origins of Cell-Free Protein Expression
The basic principle of cell-free expression has been around since the late 1800s, when it was first introduced by Eduard Buchner, a researcher who was developing the concept as a means of converting sugar to carbon dioxide and ethanol in yeast extract (1).
In the years that followed, laboratories came to adopt the technique for protein synthesis for the purposes of answering the age-old question: exactly what role do amino acids play in proteins? Scientists Marshall Nirenberg and Heinrich Matthaei made a huge breakthrough in the answer to this fundamental question in 1961, successfully applying cell-free protein expression to make the connection between nucleotide triplets and the amino acids they encode.
Using an in vitro translation system based on E. coli, they were able to synthesize the polypeptide polyphenylalanine. From there, they were able to determine the connection between the amino acid phenylalanine and its corresponding codon UUU, essentially discovering the key to cracking the genetic code. This groundbreaking experiment would eventually lead to the deciphering of all the remaining amino acid codons and laid the foundation for the wide variety of translation biology systems that are available today (2).
Basic Principles of Translation Biology
The portfolio of today’s existing systems, though extending over a broad and diverse spectrum, all fall under one of the two types of cell-free expression systems: Translation Systems and Coupled Transcription and Translation (TnT®) Systems. Despite the differences between the systems, the basic principles of the cell-free reaction remain the same.
Cell-free expression begins with crude extracts generated from cultured cells that are typically engaged in a high rate of protein synthesis, such as immature red blood cells (reticulocytes). These crude extracts are depleted of their endogenous DNA and mRNA, and the cell lysate is subsequently supplemented with macromolecular components required to perform translation, including ribosomes, tRNAs, aminoacyl-tRNA synthetases and initiation, elongation and termination factors. The process of translation is then initiated by adding a suitable template (DNA or mRNA) and carried out at an appropriate temperature. In Translation Systems, reactions are initiated with purified mRNA, while systems that are initiated with linear or plasmid DNA templates are referred to as Coupled Transcription and Translation (TnT®) Systems.
To ensure efficient translation, each extract requires additional supplementation with amino acids, energy sources (ATP, GTP), energy regenerating systems and salts (e.g., Mg2+, K+). Creatine phosphate and creatine phosphokinase typically serve as energy regenerating systems in eukaryotic systems, whereas prokaryotic systems are often supplemented with phosphoenolpyruvate and pyruvate kinase. Additionally, coupled transcription and translation systems are also typically supplied with phage-derived RNA polymerase (T3, T7 or SP6), which transcribes mRNA from an exogenous DNA template and allows for expression of genes cloned downstream of a T3, T7 or SP6 promoter.
Cell Extract and System Selection
When it comes down to selecting the right cell-free protein expression system for you, there are several factors that need to be taken into consideration, including the type of template you will be using, your desired protein yield and the intended downstream applications.
The most popular commercially-available in vitro translation systems to date consist of E. coli, wheat germ, rabbit reticulocytes or insect cell extracts. As each of these cells behave and function in different ways, the same is also true of their derived extracts, and each have their own advantages and disadvantages, which are briefly highlighted below (1; 3).
The prokaryotic E. coli system is currently the most popular protein expression system for several reasons. E. coli extraction preparation is simple and inexpensive, as E. coli is easily fermented in large quantities utilizing low-cost media and is ruptured easily using high-pressure homogenizers. E. coli-based systems also generally achieve the highest protein yields, and the total reaction cost of E. coli system is the collective lowest. E. coli is capable of activating metabolic reactions in the extract which in turn fuels high-level protein synthesis, which eliminates the need for more expensive energy substrates like phosphoenolpyruvate.
The Wheat Germ Extract (WGE), Rabbit Reticulocyte Lysate (RRL) and Insect Cell Extract (ICE) systems are currently the most widely used eukaryotic systems. These systems are advantageous in production of more complex proteins, and can also achieve post-translational modifications that are not found in E. coli . However, these eukaryotic systems do generally involve more laborious extract preparation procedures, which can increase cost. The eukaryotic systems also tend to result in lower protein yields in batch reactions when compared to E. coli systems.
Cell-free extracts of wheat germ and rabbit reticulocyte lysate support the in vitro translation of a wide variety of viral, prokaryotic and eukaryotic mRNAs. These RNA-driven systems are widely used to identify mRNA species and characterize their products. Starting with the DNA of interest, in vitro transcripts (5–80µg/ml) for translation can be generated with the RiboMAX™ Large Scale RNA Production Systems (Cat.# P1280, P1300) and the T7 RiboMAX™ Express Large Scale RNA Production System (Cat.# P1320).
E. coli S30 Extract (ECE):
Advantages: The extraction preparation is both simple and cost-effective. ECE systems have the capability of folding complex proteins, consistently have a high rate of protein synthesis and a resulting high protein synthesis yield. Additionally, the energy sources are low-cost, the current methods are well-understood and there are many well-established tools available to perform genetic modifications as well.
Disadvantages: The number of optional post-translational modifications is limited, and there are no endogenous membrane structures for synthesis of integral membrane proteins.
Wheat Germ Extract (WGE):
Advantages: A wide-spectrum expression of eukaryotic proteins have been achieved repeatedly utilizing WGE. This is also a highly productive system, which translates into a high yield of complex proteins. Sophisticated high-throughput method for proteomics.
Disadvantages: The lysate preparation can be expensive and labor-intensive. Limited post-translational modifications are possible, there are no endogenous membrane structures for synthesis of integral membrane proteins and WGE offers a low protein yield in comparison to prokaryotic systems.
Rabbit Reticulocyte Lysates (RRL):
Advantages: Cells are easy to break and the extraction preparation process is quick. The RRL system is a tried-and-true, well-established system. This system is well-suited for mammalian system eukaryotic-specific modifications, with moderate/low yields for protein.
Disadvantages: Low protein yield. Post-translational modifications only possible by supplementing with exogenous microsomal membranes.
Insect Cell Extract (ICE):
Advantages: Cells are easy to break and the extraction preparation process is quick. Many eukaryotic-specific post-translational modifications are possible using this extract, including glycosylation, disulfide-bridge formation, lipidation, and signal peptide cleavage phosphorylation. Endogenous microsomes are also available, and direct synthesis and integration of membrane proteins has been performed successfully using this extract.
Disadvantages: Insect cell extracts tend to have high cultivation costs.
Ready to start looking for your system? Explore the breadth of our Protein Expression portfolio to discover the right solution for your cell-free protein expression needs.
Advantages of Cell-Free Protein Expression Systems
Cell-free protein expression systems offer several distinct advantages in comparison to cell-based protein expression, including increased overall yields of full-length proteins that are both functional and soluble, as well as considerable time savings. Typical in vivo methods can take, at best, days and at worst, weeks, to complete (4). In stark contrast, in vitro translation reactions, including the time invested in preparation of extracts, can be performed in only a few hours, providing the fastest way to correlate phenotype (the function of expressed protein) to genotype.
Utilizing a cell-free approach, protein synthesis is also versatile in that it can be performed using a variety of inputs. Cell-free translational systems utilize mRNA as a template, while either plasmid DNA or linear PCR fragments can serve as a DNA template in coupled transcription/translation systems.
Another advantage of CFPS is that it is an open reaction. As cell-free protein expression is not confined by cell walls, there is abundant opportunity for direct manipulation of the chemical environment, allowing the addition of external components and molecules to create conditions more conducive to protein folding and activity (5). The CFPS format allows for active monitoring, rapid sampling and screening without requiring a gene cloning step (3).
As there are no cell barriers to constrain the translation control, CFPS systems are also ideal methods for synthesizing proteins that are traditionally much more difficult to express and subsequently analyze. Membrane proteins, viral proteins, toxic proteins and proteins that undergo rapid proteolytic degradation by intracellular processes can also more easily be expressed when there is no internal cell metabolism or biochemical pathways to contend with. Thus, alleviating concerns of potential toxicity of product proteins.

The wide variety of cell-free protein expression systems lends itself to a broad spectrum of protein characterization applications, from nucleic acid programmable protein array (NAPPA) to enzyme engineering using display technologies. The cell-free approach also lends itself to specific protein labeling with fluorescence, biotin, radioactivity or heavy atoms, via modified charged tRNAs or amino acids.
Eukaryotic Cell-Free Protein Expression
The eukaryotic cell-free expression systems (RRL, wheat germ or insect cell extract) are either translation systems that are primed with mRNA or coupled transcription/translation (TnT®) systems supplemented with the optimal phage RNA polymerases (T7, SP6 or T3) and primed with plasmid DNA or PCR DNA containing the T7, SP6 or T3 promoter. Coupled eukaryotic cell-free systems combine a prokaryotic phage RNA polymerase with eukaryotic extracts and utilize an exogenous DNA or PCR-generated templates with a phage promoter for in vitro protein synthesis (Figure 3).

Cell-free transcription and/or translation systems offer considerable utility, especially in functional proteomics. In particular, the recent development of the higher yield expression systems has expanded their application (6).
Cell-Free Translation Systems
The Rabbit Reticulocyte Lysate Translation Systems (Nuclease-treated and Untreated), and Wheat Germ Extract System are used for translation of mRNA. The Rabbit Reticulocyte Lysate, Nuclease-Treated (Cat.# L4960), has been optimized for mRNA translation by adding several supplements. These include hemin, which prevents activation of the heme-regulated eIF-2a kinase (HRI); an energy-generating system consisting of pretested phosphocreatine kinase and phosphocreatine; and calf liver tRNAs, to balance the accepting tRNA populations, thus optimizing codon usage and expanding the range of mRNAs that can be translated efficiently. In addition both lysates are treated with micrococcal nuclease to eliminate endogenous mRNA, thus reducing background translation. The Flexi® Rabbit Reticulocyte Lysate System (Cat.# L4540) provides greater flexibility of reaction conditions than the Rabbit Reticulocyte Lysate, Nuclease-Treated, by allowing translation reactions to be optimized for a wide range of parameters, including Mg2+ and K+ concentrations, and offers the choice of adding DTT.
In contrast to treated RRL, the Rabbit Reticulocyte Lysate, Untreated (Cat.# L4151), contains the cellular components necessary for protein synthesis (tRNA, ribosomes, amino acids, initiation, elongation and termination factors) but has not been treated with micrococcal nuclease. Untreated Rabbit Reticulocyte Lysate is not recommended for in vitro translation of specific mRNAs.
Wheat Germ Extract (Cat.# L4380) contains the cellular components necessary for protein synthesis (tRNA, ribosomes, initiation, elongation and termination factors). The extract is optimized further by the addition of an energy-generating system consisting of phosphocreatine and phosphocreatine kinase, spermidine to stimulate the efficiency of chain elongation and thus overcome premature termination, and magnesium acetate at a concentration recommended for the translation of most mRNA species. Only the addition of exogenous amino acids (including an appropriately labeled amino acid) and mRNA are necessary to stimulate translation. For further optimization, Potassium Acetate can be added for translation of a wide range of mRNAs.
Cell-Free Transcription/Translation Systems
Coupled transcription/translation systems offer researchers time saving alternative for eukaryotic in vitro transcription and translation by coupling transcription/translation into a one-tube system. Standard Rabbit Reticulocyte Lysate or Wheat Germ Extract translations (7) use RNA synthesized in vitro (8). The RNA is then used as a template for translation. Coupled systems like the TnT® Systems bypass many of these steps by incorporating the reagents needed for transcription directly in the translation mix.
In most cases, the TnT® System reactions produce significantly more protein (two- to sixfold) in a 1- to 2-hour reaction than standard in vitro Rabbit Reticulocyte Lysate or Wheat Germ Extract translations using RNA templates. In addition, TnT® Lysates also can be used with microsomal membranes to study processing events.
Microsomal vesicles are used to study co-translational and initial post-translational processing of proteins (9; 10; 11). Processing events such as signal peptide cleavage (12), membrane insertion (13), translocation and core glycosylation (14) can be examined by translation of the appropriate mRNA in vitro in the presence of microsomal membranes. Processing and glycosylation events may also be studied by transcription/translation of the appropriate DNA.
Alternatives to Rabbit Reticulocyte Lysate systems include wheat germ-based systems and systems using extracts from insect cell lines such as the commonly used Spodoptera frugiperda Sf21 cell line (15), a component of our TnT® T7 Insect Cell Extract Protein Expression System (Cat.# L1101, L1102). Wheat germ extract-based cell-free protein synthesis provides unique advantages over other cell-free lysates, including room temperature incubations, the ability to do high-throughput screening, flexibility to add auxiliary components, expression of proteins toxic to cells and screening of protein folding and function (16; 17).
TNT® Quick Coupled Transcription/Translation Systems
The components of this Master Mix have been carefully adjusted to maximize both expression and fidelity for most gene constructs. When necessary, Magnesium Acetate and Potassium Chloride can be used to optimize in vitro translation reactions with the TnT® Quick Systems.
The inclusion of RNasin® Ribonuclease Inhibitor directly in the Master Mix protects against potential disaster from the introduction of RNases carried over in the DNA solutions prepared using some miniprep protocols.
The TNT® Quick Coupled Transcription/Translation Systems
The TNT® Quick Coupled Transcription/Translation Systems provide convenient single-tube, coupled transcription/translation reactions for eukaryotic cell-free protein expression.
Learn MoreProkaryotic Cell-Free Protein Expression
E. coli S30 Extract Systems
Typically, E. coli S30 fraction is used for prokaryotic expression. Although the choice of systems should not be determined just by the origin of the target protein, but also by the biological nature of the protein and the requirements of downstream applications. Yields from E.coli-based systems can be much greater than eukaryotic-based systems, often as high as a few mg/mL depending on protein and reaction format.
The S30 T7 High-Yield Protein Expression System (Cat.# L1110, L1115) is an E. coli extract-based cell-free protein synthesis system. It simplifies the transcription and translation of DNA sequences cloned in plasmid containing a T7 promoter by providing an extract that contains T7 RNA polymerase for transcription and all necessary components for translation. This system can produce high levels of recombinant proteins (up to hundreds of micrograms of recombinant protein per milliliter of reaction) within an hour using a vector containing the sequence of interest, a T7 promoter and a ribosome-binding site (RBS).
The S30 extracts in the E. coli S30 Extract Systems are prepared by modifications of the methods described by Zubay (18; 19; 20). For expression using linear templates (Cat.# L1030), the extract is prepared from E. coli B strains deficient in ompT endoproteinase and ion protease activity. This results in greater stability of expressed proteins, which would otherwise be degraded by proteases if expressed in vivo (21; 22). When circular template DNA is used, E. coli S30 Extract Systems (Cat.# L1020) can produce higher expression levels of proteins that are normally expressed at low levels in vivo due to the action of host-encoded repressors (23). An optimized S30 Premix Plus provides all other components required to express high levels of recombinant proteins.
Proteins expressed in the E. coli S30 Extract Systems may be used for a variety of functional transcription and translation studies. The most common application of E. coli S30 Extract Systems is the synthesis of small amounts of radiolabeled protein for use as a tracer in protein purification, and for incorporation of unnatural amino acids into proteins for structural studies (24).
Applications of Cell-Free Protein Expression Systems
As our knowledge of cell-free expression systems and their capabilities has continuously expanded, researchers have been able to exploit the various advantages of these systems to develop novel protein technologies. We explore some of these unique applications below.
Functional Genome and Proteome Analysis
Cell-free protein synthesis provides a basic, straightforward tool to aid in the identification of protein and molecular interactions, such as protein-protein, protein-nucleic acid, and protein-ligand. In order to characterize these interactions, one of the desired entities (nucleotide/ligand/protein etc.) is labelled, then incubated in a CFPS system with the other desired entity. The resulting complex is then either isolated utilizing a technique like immunoprecipitation, or detected directly by a electrophoretic mobility-shift assay (EMSA), in which protein interaction complexes are slowed when compared to their unbound counterparts (5).
Cell-free protein expression systems are also valuable tools for manufacturing protein arrays. Protein microarrays are solid-phase ligand binding assay systems that are utilized to track protein activity and interaction, and to determine protein function in a high-throughput format. Traditional cell-based generation of these protein chips is a labor-intensive process which requires expression and purification of each individual protein to be arrayed. Another drawback is long-term functional stability of the immobilized proteins is usually limited. The use of CFPS can circumvent these challenges, through the parallel synthesis of multiple proteins directly in situ (on-chip).
There are several different types of protein microarrays being utilized in current research, including PISA (Protein In Situ Array), NAPPA (Nucleic Acid Programmable Protein Array) and DAPA (DNA Array to Protein Array) .
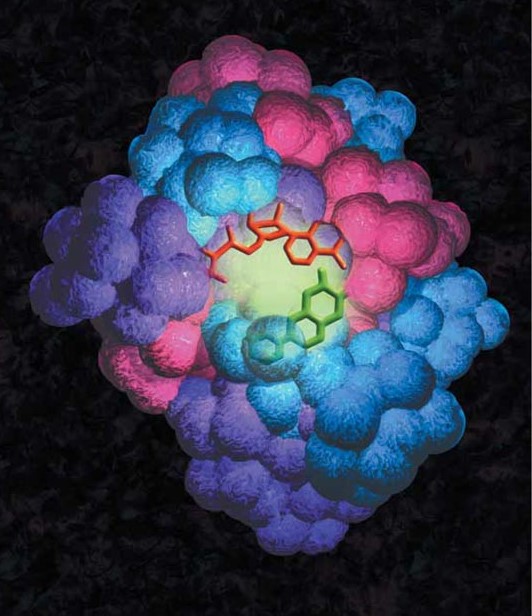
PISA
The PISA method, the first well-known cell-free in situ protein microarray technology, involves the rapid production of tagged proteins directly from DNA fragments by cell-free expression in solution. The individual DNA constructs that encode for the protein of interest contain a T7 promoter, sequences for translation initiation and termination, as well as an N- or C-terminal tag sequence. The DNA constructs are generated via PCR or RT-PCR utilizing a high fidelity TAQ polymerase, then cell-free coupled transcription and translation expresses the desired tagged proteins (25). Following synthesis, the proteins are simultaneously captured and immobilized in situ, by the tag-capturing coating on the surface of the slide (26).
NAPPA
NAPPA is a label-based technology, in which the DNA template is biotinylated and immobilized onto a slide pre-coated with avidin and an anti-GST antibody that functions as the protein capture reagent (27). The array can then be used for to express targeted proteins in situ by using rabbit reticulocyte lysate or similar CFPS system. Following translation, the targeted proteins are captured by the immobilized antibody in each spot, resulting in a protein array in which each protein is co-localized with its corresponding expression plasmid (28).
The NAPPA method has demonstrated particular value in the characterization of disease biomarkers and study of autoimmune disease. NAPPA-generated microarrays have been used to help identify new antibody responses to the Mycobacterium tuberculosis proteome, effectively pinpointing 8 proteins with tuberculosis biomarker value (29). NAPPA has also been utilized to characterize autoantibody response in patients with the autoimmune rheumatic disease Ankylosing spondylitis (30), to profile circulating and synovial antibodies in patients with juvenile idiopathic arthritis (31), and to detect antibodies that bind tumor antigens in breast cancer (32).
DAPA
The DNA array to protein array (DAPA) method was developed as a means of producing protein arrays on demand by printing them from a single DNA array template. In this method, a slide containing an array of immobilized PCR-amplified fragments encoding for a set of tagged proteins is assembled face-to-face with a second slide, pre-coated with a protein tag-capturing reagent. A permeable membrane containing a cell-free lysate is then sandwiched between the two slide surfaces, enabling coupled transcription and translation. Protein synthesis originates from the spots of the immobilized DNA, and the synthesized proteins diffuse through the membrane and are immobilized on the capture slide surface, creating the protein array (33).
Expression of Difficult-to-Express Proteins
Cell-free protein expression systems provide a means to produce functional proteins that are typically difficult to express, such as membrane proteins, toxic proteins and viral proteins. Using a coupled CFPS system, a complex heterotetrameric and multiple disulfide-bridged IgG molecule has been assembled successfully and completely under suitable oxidation and folding conditions (34). Various cell-free systems have also successfully been used to synthesize membrane proteins in vitro, including G-protein-coupled receptors, epidermal growth factor receptor and ATP synthase (35).
In vitro translation systems have also been used to successfully express viral proteins, virus-like particles (VLPs) and vaccine antigens. In one study, 124 P. falciparum genes of interest were synthesized in vitro as potential malaria vaccine candidates, resulting in the successful synthesis of 75% of the products in soluble form, without the need for codon optimization. Cell-free E. coli extract systems have been applied to synthesize both VLPs and vaccines, including anti-influenza VLPs, a B-cell lymphoma vaccine and anti-hepatitis B VLPs (35).
In vitro systems have effectively produced infectious encephalomyocarditis virus (EMCV), and the entire open reading frame of hepatitis C virus RNA has been correctly translated and processed when supplemented with canine microsomal membranes (5). These studies demonstrate the enormous potential of cell-free expression systems not only as valuable tools for understanding the mechanisms of viral replication, but also as a means for vaccine discovery and prospective platform for large-scale production of vaccines (5; 35).
Protein Evolution and Enzyme Engineering
Proteins are complex, versatile molecules that serve a variety of functions in the cell. Directed protein evolution is a cyclic process of alternating between the selection of functional gene variants and gene diversification, that can be utilized as a means of improving certain types of protein functions, like catalytic activity, binding affinity and thermostability (4; 36). Several unique protein technologies in directed evolution, including ribosome display, mRNA display and in vitro compartmentalization, have been developed in the last thirty years using in vitro translation systems. In comparison to cell-based methods, these in vitro technologies offer several advantages, including efficient linking of genetic information (genotype) to their encoded proteins (phenotype) and broader range of library sizes.
Ribosome Display
The ribosome display method involves isolating individual proteins from polypeptide DNA sequence libraries. During in vitro translation, protein-ribosome-mRNA (PRM) complexes are established by connecting the nascent polypeptide to its encoding mRNA (1).
This PRM of interest can then be captured through affinity binding via immobilized ligand, from which the mRNA can then be recovered and reverse transcribed back into cDNA to be mutated as desired. This process can also be repeated without mutating the recovered cDNA as a means of enriching target proteins from large populations (5).
The ribosome display technology (Figure 6) has been a popular method for in vitro selection and evolution of antibodies, as well as transcription factors, receptors, proteases, enzymes, novel peptides, tag sequences and ligand-binding domains and motifs (5). Ribosome display has also been utilized in proteomics applications, leading to the identification of a sequence of 5'-untranslated region that promotes translation efficiency (37), as well as comprehensive determination of antigenic proteins for a cDNA library of Staphylococcus aureus that could be used as potential vaccine candidates (38).
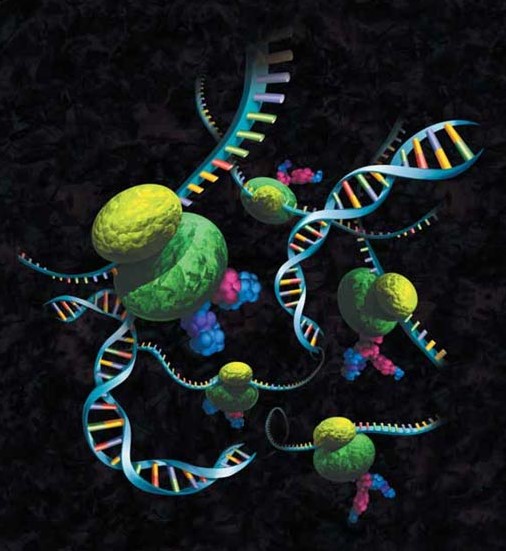
mRNA Display
In mRNA display technology, a large DNA library encoding the polypeptide of interest is transcribed into mRNA. The 3’ end of mRNA is ligated to a short, single-strand DNA oligonucleotide which carries an adaptor molecule, typically puromycin. The modified mRNA product is translated via cell-free protein synthesis; the puromycin enters the ribosome and forms a peptide bond with the nascent polypeptide chain. Then cDNA is generated via reverse transcription to stabilize the nucleic acid component and facilitate recovery of the genetic information following selection. This cDNA can then be amplified via PCR for further study or other use (39).
This mRNA display technology has been employed in the successful directed evolution of ligases as well as ATP-binding domains from artificial random-sequence libraries (40; 41). mRNA display has also been applied to obtain antigen-binding proteins that were based on the scaffold fibronectin type III, resulting in production of high-affinity molecules capable of binding TNF-alpha (42) and recognition of specific endogenous phosphorylated IkBalpha (43).
In Vitro Compartmentalization
In vitro compartmentalization (IVC) is a method of directed evolution where the objective is to split up a large reaction into many microscopic compartments, which encapsulate DNA and its associated mRNA and protein(s), in either water-in-oil emulsion microdroplets or on the surface of microbeads. Each of these droplets contain a single gene, in addition to all the necessary molecular components required, which are then transcribed and translated. Proteins that express the desired activity convert substrate into a product that will remain linked to the gene, as they are completely confined to the microdroplet. Genes that are linked to the product are either selectively enriched, amplified and characterized, or linked back to the substrate and subjected to additional rounds of selection via compartmentalization. The IVC strategy has successfully been applied in the engineering of several enzymes, including methyltransferases, polymerase and restriction endonucleases (5; 44).
Screenings
Incorporating cell-free expression systems into high-throughput screening applications enables both in situ and on-demand expression of proteins and peptides. These cell-free expression high-throughput screening applications can be categorized broadly into on-chip technologies and in vitro displays, several selection methods previously discussed in the Protein Evolution and Enzyme Engineering section.
In the in vitro display selection technologies, the expressed complexes are directly screened and tested for activity, and the linked genetic sequence is used in successive rounds of enrichment. On-chip selection technologies immobilize expressed proteins in treated surfaces without their source DNA or RNA (45).
There are a variety of high-throughput screening methods currently available, all of which are capable of streamlining the traditional screening process. These screening methods include microtiter plates, digital imaging, fluorescence-activated cell sorting (FACS), cell surface display, IVTC and resonance energy transfer (46).
In vitro translation followed by screening has largely been employed in medical applications, enabling the development of the Protein Truncated Test (PTT) (Figure 7) which utilizes open reading frames of genetic diseases caused by translation-terminating mutations as diagnostics (5). Cell-free protein expression systems have also enabled screening of toxic reagents (47) and identification of translation inhibitors as potential drugs (48). Screening and production of potential vaccine candidates via in vitro protein expression led to the identification of some effective vaccines that protected mice against tumors with the same efficacy as those produced in a mammalian cell (49).
Protein Labeling and Detection
Detection of proteins expressed using cell-free systems is necessary for most applications such as protein:protein interaction and protein:nucleic acid interaction studies. Traditionally, radioactive [35S]methionine has been added to cell-free expression reactions, and the methionine is incorporated into the expressed protein, allowing detection by autoradiography. Many researchers are moving away from radioactivity due to high costs, regulations, radioactive exposure and waste disposal issues. Traditional Western blot analysis provided researchers with a non-radioactive method for detection but, if performed improperly, could result in high backgrounds. However, detection methods such as FluoroTect™ GreenLys in vitro Translation Labeling System (Cat.# L5001) and the Transcend™ Non-Radioactive Translation Detection System (Cat.# L5070, L5080) allow Western blotting with sensitive detection and low backgrounds (50).
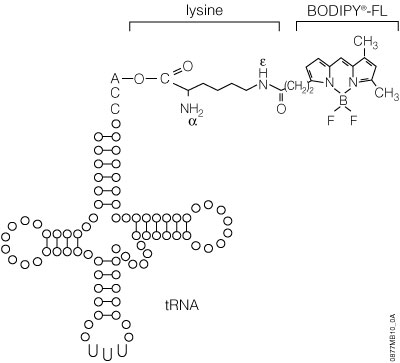
The FluoroTect™ System employs a tRNA charged with a lysine that is labeled at the e position with the BODIPY®-FL fluorophore. These fluorescently labeled lysine residues are incorporated into synthesized proteins during in vitro translation. The Transcend™ System relies on incorporation of biotinylated lysine residues into nascent proteins during translation. The biotinylated lysine is added to the translation reaction as a charged e-labeled biotinylated-lysine:tRNA complex (Transcend™ tRNA) rather than a free amino acid. After SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotting, biotinylated proteins can be visualized by binding Streptavidin-AP or Streptavidin-HRP, followed by colorimetric or chemiluminescent detection, respectively. Typically, these methods can detect 0.5–5ng of protein, with a sensitivity equivalent to that achieved with [35S]methionine incorporation and autoradiographic detection.
Transcend™ Non-Radioactive Translation Detection Systems
The Transcend™ Non-Radioactive Translation Detection Systems enable non-radioactive detection of proteins synthesized in vitro. Using this system, biotinylated lysine residues are incorporated into nascent proteins during translation, eliminating the need for labeling with [35S]methionine or other radioactive amino acids. Biotinylated lysine is added to the translation reaction as a pre-charged e-labeled biotinylated lysine-tRNA complex (Transcend™ tRNA) rather than a free amino acid. After SDS-PAGE and electroblotting, the biotinylated proteins can be visualized by binding either Streptavidin-Alkaline Phosphatase (Streptavidin-AP) or Streptavidin-Horseradish Peroxidase (Streptavidin-HRP), followed either by colorimetric or chemiluminescent detection. Typically, 0.5–5ng of protein can be detected within 3–4 hours after gel electrophoresis. This sensitivity is equivalent to that achieved with [35S]methionine incorporation and autoradiographic detection 6–12 hours after gel electrophoresis. For a detailed protocol and background information, please see Technical Bulletin #TB182.
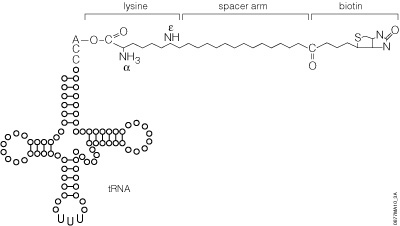
The use of Transcend™ tRNA offers several advantages:
- No radioisotope handling, storage or disposal is needed.
- The biotin tag allows detection (0.5–5ng sensitivity).
- The biotin tag is stable for 12 months, both as the Transcend™ tRNA Reagent and within the labeled proteins. It is not necessary to periodically resynthesize biotin-labeled proteins, unlike [35S]-labeled proteins, whose label decays over time.
- Labeled proteins are detected as sharp gel bands, regardless of the intensity of labeling or amount loaded on the gel, thus allowing the detection of poorly expressed gene products.
- Results can be visualized quickly, using either colorimetric or chemiluminescent detection.
The precharged E. coli lysine tRNAs provided in this system have been chemically biotinylated at the e-amino group using a modification of the methodology developed by Johnson et al. (1976). The biotin moiety is linked to lysine by a spacer arm, which greatly facilitates detection by avidin/streptavidin reagents (Figure 8). The resulting biotinylated lysine tRNA molecule (Transcend™ tRNA) can be used in either eukaryotic or prokaryotic in vitro translation systems such as the TNT® Coupled Transcription/Translation Systems, Rabbit Reticulocyte Lysate, Wheat Germ Extract or E. coli S30 Extract (52). Lysine is one of the more frequently used amino acids. On average, lysine constitutes 6.6% of a protein’s amino acids, whereas methionine constitutes only 1.7% (53).
Effects of Biotinylated Lysine Incorporation on Expression Levels and Enzyme Activity
Lysine residues are common in most proteins and usually are exposed at the aqueous-facing exterior. The presence of biotinylated lysines may or may not affect the function of the modified protein. In gel shift experiments, c-Jun synthesized in TNT® Reticulocyte Lysate reactions and labeled with Transcend™ tRNA performed identically to unlabeled c-Jun (54).
Estimating Incorporation Levels of Biotinylated Lysine
Incorporation of radioactively labeled amino acids into proteins typically is quantitated as percent incorporation of the label added. This value can include incorporation of radioactivity into spurious gene products such as truncated polypeptides. Thus, percent incorporation values provide only a rough estimate of the amount of full-length protein synthesized and do not provide any information on translation fidelity. With Transcend™ tRNA reactions, it is difficult to directly determine the percent incorporation of biotinyl-lysines into a translated protein. An alternative means of estimating translation efficiency and fidelity in Transcend™ tRNA reactions is to determine the minimum amount of products detectable after SDS-PAGE. In all cases tested, we detected translation products in 1µl of a 50µl translation reaction using as little as 0.5µl of Transcend™ tRNA (Figure 9). The amount of biotin incorporated increases linearly with the amount of Transcend™ tRNA added to the reaction, up to a maximum at approximately 2µl.
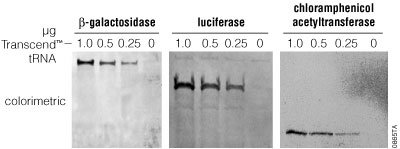
Capture of Biotinylated Proteins
Biotinylated proteins can be removed from the translation reaction using biotin-binding resins such as SoftLink™ Soft Release Avidin Resin (Cat.# V2011, V2012). Nascent proteins containing multiple biotins bind strongly to SoftLink™ Resin and cannot be eluted using “soft-release” nondenaturing conditions. SoftLink™ Resin is useful, however, as a substitute for immunoprecipitation.
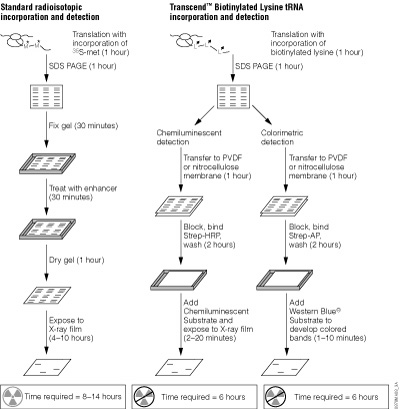
Colorimetric and Chemiluminescent Detection of Translation Products
Biotin-containing translation product can be analyzed in either of two ways. The product can be resolved directly by SDS-PAGE, transferred to an appropriate membrane and detected by either a colorimetric or chemiluminescent reaction (Figure 10). Alternatively, biotinylated protein can be captured from the translation mix using a biotin-binding resin such as SoftLink™ Resin. This approach is useful as a replacement for immunoprecipitation of protein complexes.
The FluoroTect™ GreenLys in vitro Translation Labeling System
The FluoroTect™ GreenLys in vitro Translation Labeling System uses a charged lysine tRNA molecule labeled with the fluorophore BODIPY®-FL at the epsilon (e) amino acid position of lysine (Figure 11). For the FluoroTect™ System, lysine was chosen as the labeled amino acid because it is one of the more frequently used amino acids, comprising, on average, 6.6% of a protein’s amino acids. Detection of the labeled proteins is accomplished in 2–5 minutes directly “in-gel” by use of a laser-based fluorescent gel scanner. This eliminates any requirement for protein gel manipulation such as fixing/drying or any safety, regulatory or waste disposal issues such as those associated with the use of radioactively labeled amino acids. The convenience of non-isotopic “in-gel” detection also avoids the time-consuming electroblotting and detection steps of conventional non-isotopic systems. For a detailed protocol and background information about this system, please see Technical Bulletin #TB285.
References
-
Zahnd, C. et al. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods. 4(3), 269–79.
- Ginsburg, J. (2009) Deciphering the Genetic Code: A National Historic Chemical Landmark. This can be viewed online at: https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/geneticcode.html#poly-u-experiment.
- Carlson, E. et al. (2012) Cell-Free Protein Synthesis: Applications Come of Age. Biotechnol. Adv. 30(5), 1185–1194.
- Chong, S. (2014) Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial Systems, and Expanding Applications. Curr. Protoc. Mol. Biol. 108, 16.30.1–11.
- He, M. (2008) Cell-free protein synthesis: applications in proteomics and biotechnology. New Biotech. 25(2–3), 126–132.
- Hurst, R. (2011) Innovative Applications for Cell-Free Expression. Promega Corporation Website, accessed June 2019.
- Pelham, H.R.B. and Jackson, R.J. (1976) An efficient mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem. 67, 247–56.
- Krieg, P. and Melton, D. (1984) Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucl. Acids Res. 12, 7057–70.
- Rando, R.R. (1996) Chemical biology of protein isoprenylation/methylation. Bichimica Biophysica Acta. 1300, 5–16
- Han K.-K. and Martinage, A. (1992) Post-translational chemical modification(s) of proteins. Int. J. Biochem. 24, 19–28.
- Chow, M. et al. (1992) Structure and biological effects of lipid modifications on proteins. Curr. Opin. Cell Biol. 4, 629–36.
- MacDonald, M.R. et al. (1988) Posttranslational processing of alpha-, beta-, and gamma-preprotachykinins. Cell-free translation and early posttranslational processing events. J. Biol. Chem. 263, 15176–83.
- Ray, R.B. et al. (1995) Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein. Virus Research. 37, 209–20.
- Bocco, J.L. et al. (1988) Processing of SP1 precursor in a cell-free system from poly(A+) mRNA of human placenta. Mol. Biol. Reports. 13, 45–51.
- Ezure, T. et al. (2006) Cell-free protein synthesis system prepared from insect cells by freeze-thawing. Biotechnol. Prog. 22, 1570–7.
- Morita, E.H. et al. (2003) A wheat germ cell-free system is a novel way to screen protein folding and function. Protein Sci. 12, 1216–21.
- Vinarov, D.A. et al. (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nature Methods. 12, 149–53.
- Zubay, G. (1973) In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7, 267–87.
- Zubay, G. (1980) The isolation and properties of CAP, the catabolite gene activator. Meth. Enzymol. 65, 856–77.
- Lesley, S.A. et al. (1991) Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J. Biol. Chem. 266, 2632–8.
- Pratt, J.M. (1984) In: Transcription and Translation, Hanes, B.D. and Higgins, S.J., eds., IRL Press, Oxford, UK.
- Studier, F.W. and Moffatt, B.A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes.J. Mol. Biol. 189, 113–30.
- Collins, J. (1979) Cell-free synthesis of proteins coding for mobilisation functions of ColE1 and transposition functions of Tn3. Gene.6, 29–42.
- Noren, C.J. et al. (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 244, 182–8.
- Nand, A. et al. (2012) Emerging technology of in situ cell free expression protein microarrays. Protein & Cell. 3(2), 84–88.
- He, M. and Taussig, M.J. (2001) Single step generation of protein arrays from DNA by cell-free expression and in situ immobilization (PISA method). Nucleic Acids Res. 29 (15), e73.
- Manzano-Román, R. and Fuentes, M. (2019) A decade of Nucleic Acid Programmable Protein Arrays (NAPPA) availability: News, actors, progress, prospects and access. J. Proteomics. 198, 27–35.
- Berrade, L. et al. (2011) Protein Microarrays: Novel Developments and Applications. Pharm. Res. 28(7), 1480–1499.
- Song, L. et al. (2017) Identification of antibody targets for tuberculosis serology using high-density nucleic acid programmable protein arrays. Mol. Cell. Proteom. 16, 277–289.
- Wright, C. et al. (2012) Detection of Multiple Autoantibodies in Patients with Ankylosing Spondylitis Using Nucleic Acid Programmable Protein Arrays. Mol. Cell. Proteomics.11(2), M9.00384.
- Gibson, D. et al. (2012) Circulating and synovial antibody profiling of juvenile arthritis patients by nucleic acid programmable protein arrays. Arthritis Research & Therapy. 14, R77.
- Anderson, K.S. et al. (2008) Application of protein microarrays for multiplexed detection of antibodies to tumor antigens in breast cancer. J. Proteome Res., 7, 1490–1499.
- He, M. et al. (2008) Printing protein arrays from DNA arrays. Nature Methods. 5, 175–177.
- Frey, S. et al. (2007) Synthesis and characterization of a functional intact IgG in a prokaryotic cell-free expression system. Biol. Chem.389, 37–45.
- Perez, J. et al. (2016) Cell-Free Synthetic Biology: Engineering Beyond the Cell. Cold Spring Harb. Perspect. Biol. 8(12), a023853.
- Packer, M. and Liu, D. (2015) Methods for the directed evolution of proteins. Nature Reviews Genetics. 16, 379–394.
- Mie, M. et al. (2008) Selection of mRNA 5’-untranslated region sequence with high translation efficiency through ribosome display. Biochem. Biophys. Res. Commun. 373, 48–52.
- Weichhart, T. et al. (2003) Functional selection of vaccine candidate peptides from Staphylococcus aureus whole-genome expression libraries in vitro. Infect. Immun. 71, 4633–4641.
- Lipovsek, D. and Plückthun, A. (2004) In-vitro protein evolution by ribosome display and mRNA display. J. Imm. Methods. 290(1–2), 51–67.
- Keefe, A.D. and Szostak, J.W. (2001) Functional proteins from a random-sequence library. Nature. 410, 715–718.
- Seelig, B. and Szostak, J.W. (2007) Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature. 448, 828–831.
- Xu, L. et al. (2002) Directed evolution of high affinity antibody mimics using mRNA display. Chem. Biol.9, 933–942.
- Olson, C.A. et al. (2008) mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin. ACS Chem. Biol. 3(8), 480–5.
- Miller, O. et al. (2006) Directed evolution by in vitro compartmentalization. Nature Methods. 3, 561–570.
- Contreras-Llano, L. and Tan, C. (2018) High-throughput screening of biomolecules using cell-free gene expression systems. Synth. Bio. 3(1), ysy012.
- Xiao, H. et al. (2015) High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res. 54(16), 4011–4020.
- Wei, Q. et al. (2005) Toxin detection by a minisaturised in vitro protein expression array. Anal. Chem. 77, 5494–5500.
- Brandi, L. et al. (2007) Review: methods for identifying compounds that specifically target translation. Methods Enzymol.431, 229–267.
- Kanter, G. et al. (2007) Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 109, 3393–3399.
- Hook, B. (2011) Non-Radioactive Detection of Proteins Expressed in Cell-Free Expression Systems. Promega Corporation Website, accessed June 2019.
- Johnson, A.E. et al. (1976) Nepsilon-acetyllysine transfer ribonucleic acid: A biologically active analogue of aminoacyl transfer ribonucleic acids. Biochem. 15, 569–75.
- Kurzchalia, T.V. et al. (1988) tRNA-mediated labelling of proteins with biotin. A nonradioactive method for the detection of cell-free translation products. Eur. J. Biochem. 172, 663–8.
- Dayhoff, M.O. (1978) In: Atlas of Protein Sequence and Structure, Suppl. 2 National Biomedical Research Foundation, Washington, DC.
- Crowley, K.S. et al. (1993) The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell. 73, 1101–15.