For ordering information on the products discussed here, please visit the Restriction Enzymes product listing.
History
Restriction enzymes recognize short DNA sequences and cleave double-stranded DNA at specific sites within or adjacent to these sequences. Approximately 3,000 restriction enzymes, recognizing over 230 different DNA sequences, have been discovered. They have been found mostly in bacteria, but have also been isolated from viruses, archaea and eukaryotes. It has been estimated that 25% of all bacteria contain at least one restriction enzyme (1) and as many as 7 have been found in a single species (2) .
In the early 1950s, Luria and colleagues (3,4) reported a phenomenon known as host-controlled restriction modification. They observed that bacteriophage that grew well in one bacterial strain often grew poorly in a second, forming only a few plaques. Phage isolated from these plaques were able to re-infect the second strain and grow well, but lost the ability to grow on the original strain.
Arber and Dussoix (5,6) proposed a molecular model to explain host-controlled restriction modification. They postulated that certain bacterial strains contain an endonuclease that is able to cleave DNA, and that some strains contain a strain-specific modification system that is responsible for protecting host DNA from the action of its own endonuclease. Unmodified (foreign) DNA, such as that of an infecting phage, is degraded by the endonuclease, restricting phage infection (hence the term restriction endonuclease). However, a small proportion of the phage DNA is modified prior to degradation by the endonuclease. This modified DNA is able to successfully replicate and infect the second host, but since that host does not contain the same modification system as the first, the modified phage lose their ability to replicate on the original host.
In 1968, Arber and Linn demonstrated nuclease activity of Eco B restriction enzyme (7) and Meselson and Yuan purified a similar enzyme from E. coli K (8). These were later classified as Type I restriction enzymes, which cleave DNA at random positions, often far removed from the recognition site.
In 1970, Smith and colleagues described the purification of the first Type II restriction enzyme, Hind II (9), and the characterization of its recognition and cleavage site (10). Werner Arber, Hamilton O. Smith and Daniel Nathans shared the 1978 Nobel Prize for Medicine and Physiology for their discovery of restriction enzymes and their application to molecular genetics. Because of the ability of these enzymes to cleave DNA at specific recognition sites, they have continued to play a fundamental role in cloning and DNA typing applications.
References
- Roberts, R.J. and Halford, S.E. (1993) In: Nucleases, Second Edition Linn, S.M., Lloyd, S.R. and Roberts, R.J., eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- Stein D.C. et al. (1995) Restriction and modification systems of Neisseria gonorrhoeae. Gene 157, 19–22.
- Luria, S.E. and Human, M.L. (1952) A nonhereditary, host-induced variation of bacteria viruses. J. Bacteriol. 64, 557–69.
- Bertani, G. and Weigle, J.J. (1953) Host controlled variation in bacterial viruses. J. Bacteriol. 65, 113–21.
- Arber, W. and Dussoix, D. (1962) Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda. J. Mol. Biol. 5, 18–36.
- Dussoix, D. and Arber, W. (1962) Host specificity of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda. J. Mol. Biol. 5, 37–49.
- Linn, S. And Arber, S. (1968) A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. Proc. Natl. Acad. Sci. USA 59, 1300.
- Meselson, M. And Yuan, R. (1968) DNA restriction enzyme from E. coli. Nature 217, 1110–4.
- Smith, H.O. and Wilcox, K.W. (1970) A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J. Mol. Biol. 51, 379–91.
- Kelly, T.J., Jr., and Smith, H.O. (1970) A restriction enzyme from Hemophilus influenzae. II. J. Mol. Biol. 51, 393.
Types, Definitions and Genomic Organization
A. Restriction Enzyme Classification
Restriction endonucleases are categorized into one of four general groups (Types I, II, III, and homing endonucleases based on their subunit structure, cofactor requirements, specificity of cleavage, and associated methylase activity (Table 1.2). References 1-10 provide reviews of each restriction enzyme type as follows: Type II and Type II subclasses (1)(2)(3), Type IIb (4) (5), Type IIe (6)(7), Type IIs (8), homing endonucleases (9), and Type I and Type III (10).
B. Restriction/Modification Systems
Restriction endonucleases of Types I, II and III have companion methylase(s) that recognize the same sequence as the endonuclease and methylate each strand at a specific base and position, resulting in either 4-methylcytosine, 5-methylcytosine, 5-hydroxymethylcytosine, or 6-methyladenine. Once methylated, the host DNA is no longer a substrate for the endonuclease. Hemi-methylated DNA, such as after a fresh round of replication, is also protected from digestion. The restriction endonuclease and modification methylase genes lie adjacent to each other on the host chromosomal or plasmid DNA and may be oriented transcriptionally in a convergent, divergent, or sequential manner. Occasionally, in convergent or divergent gene organization, an open reading frame encoding a regulator of endonuclease expression, often referred to as the control or "C" gene, exists immediately upstream of the endonuclease gene. As the proximity of the endonuclease and methylase genes appears to be universal, they are frequently referred to as restriction/modification (R/M) systems (11). Type III enzymes use a modified host protection mechanism (12) (13). Homing endonucleases, which are encoded by mobile, self-splicing introns or inteins, have no associated methylases.
C. Recognition Sequences
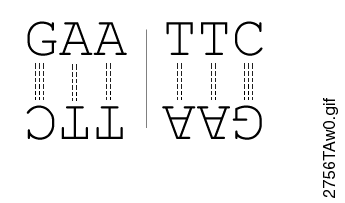
Most restriction endonucleases recognize palindromic or partially palindromic sites. A palindrome is defined as dyad symmetry around an axis. For example, EcoRI:
A set of single letter codes have been accepted for the degeneracy of partial palindromes as follows:
| R = A or G | K = G or T | S = G or C |
| Y = C or T | M = A or C | W = A or T |
| B = not A (C or G or T) | H = not G (A or C or T) | N = any nucleotide |
| D = not C (A or G or T) | V = not T (A or C or G) |
The recognition site for StyI is listed as CCWWGG. Therefore, the substrate sequences for StyI can be palindromic (CCTAGG or CCATGG) or partially palindromic (CCTTGG or CCAAGG). This flexibility or ambiguity of recognition is not currently understood. Situations where allowed nucleotides can be either purine or pyrimidine or when only a single nucleotide is excluded are particularly interesting. Interrupted palindromes may contain from 1 to 9 unspecified nucleotides between the required flanking nucleotides. Bipartite recognition sequences are interrupted but without palindromic symmetry in the specified nucleotides. Non-palindromic generally refers to uninterrupted sequences without symmetry or, at most, a single unspecified nucleotide within the sequence. Cleavage typically occurs within the recognition site except for Types I, IIb, IIs, and III. When cleaving outside the recognition sequence, the cut site is often given by the notation (N)x where X is the number of unspecified nucleotides between the 3´ end of the recognition sequence for that strand and the cut site. If only a single strand is given followed by (X/Y), X has the same meaning as before and Y is the number of unspecified nucleotides between the 5´ end of the recognition sequence and the cut site for the complementary strand. Isoschizomers are endonucleases that recognize the same sequence and cleave at the same position. Neoschizomers recognize the same sequence but cleave at different positions within that sequence.
D. Types and General Properties of Restriction Endonucleases
The table below gives the types and general properties restriction endonucleases. The sequence of the top strand is given from 5´ to 3´. Arrows indicate cleavage. In general, when the recognition site is palindromic there is a single monomeric companion methylase. For BcgI, the only Type IIb enzyme for which a structure has been proposed, the methylation activity is contained in the same subunit as the restriction activity within the heterotrimer (4). AdoMet, also referred to as S-adenosyl methionine, or SAM, is always required for methylation. For non-palindromic recognition sites, there may be one or two (strand specific) monomeric companion methylases. The intron or intein encoded enzymes have no associated methylase.
Types and General Properties of Restriction Endonucleases |
| Type II (EC 3.1.21.4) |
|
Recognition Sequence: Palindromic or interrupted palindrome, ambiguity may be allowed(4) Subunit Structure(1) (Restriction Activity): Homodimer(3) (2 R-S) Cofactors(2) and Activators: Mg2+ Cleavage Site: Defined, within recognition site, may result in a 3´ overhang, 5´ overhang, or blunt end. Example: EcoRI: G/AATT C Example(s): EcoRI, BamHI, HindIII, KpnI, NotI, PstI, SmaI, XhoI |
| Type IIb |
|
Recognition Sequence: Bipartite, interrupted Subunit Structure Restriction Activity): Heterotrimer (2 R-M, 1 S) Cofactors and Activators: Mg2+, AdoMet (for methylation) Cleavage Site: Cuts both strands on both sides of recognition site a defined, symmetric, short distance away and leaves 3´ overhangs. Example: BcgI: /10(N)CGA(N)6TCG(N)12/ Example(s): BcgI, Bsp24I, BaeI, CjeI, CjePI |
| Type IIe(5) |
|
Recognition Sequence: Palindromic, palindromic with ambiguities, or non-palindromic Subunit Structure (Restriction Activity): Homodimer (2 R-S) or monomer (R-S), similar to Type II or Type IIs Cofactors and Activators: Mg2+, also a second recognition site, acting in cis or trans binds to the endonuclease as an allosteric effector (link to glossary definition) Cleavage Site: Cuts in a defined manner within the recognition site or a short distance away. Activator DNA may be required for complete cleavage. Example: NaeI: GCC/GGC Example(s): NaeI, NarI, BspMI, HpaII, SalI, EcoRII, Eco57I(6), AtuBI, Cfr9I, SauBMKI, Ksp632I |
| Type IIs(5) |
|
Recognition Sequence: Non-palindromic, nearly always contiguous and without ambiguities Subunit Structure (Restriction Activity): Monomeric (R-S) Cofactors and Activators: Mg2+ Cleavage Site: Cuts in a defined manner with at least one cleavage site outside of the recognition sequence. Rarely leaves blunt ends. GGATG(N)9/ Example(s): FokI, Alw26I, BbvI, BsrI, EarI, HphI, MboII, SfaNI, Tth111I |
| Type: Intron or Intein encoded |
|
Recognition Sequence: 12-40bp, tolerance for base pair substitutions exists Subunit Structure (Restriction Activity): Monomer, homodimer, other protein or RNA may be required Cofactors and Activators: Mg2+, may also bind Zn2+ Cleavage Site: Leave 3´ and 5´ overhangs of 1-10 bases. A few sites have not yet been determined. One strand may be cleaved preferentially, or may be cleaved in the absence of Mg2+. Some enzymes only cleave one strand. Example (cleaving both strands): I-PpoI. CTCTC TTAA/GGTAGC Example(s): I-PpoI, I-CeuI, I-DmoI, I-SceI, PI-SceI, PI-PspI |
|
Type I and Type III Enzymes. The enzymes listed below are not commercially available at this time. The number of known Type I and Type III enzymes are quite limited and all members are listed. Both types also require ATP. There are several possibilities for the companion methylase subunit structure of these two types. |
| Type I (EC 3.1.21.3) |
|
Recognition Sequence: Bipartite, interrupted Subunit Structure(Restriction Activity): Usually a pentameric complex (2 R, 2 M, and 1 S) Cofactors and Activators: Mg2+, AdoMet, ATP (hydrolyzed) Cleavage Site: Distant and variable from recognition site. Example: EcoKI: AAC(N6)GTGC(N>400)/ Example(s): EcoKI, EcoAI, EcoBI, CfrAI, StyLTII, StyLTIII, StySPI |
| Type: III (EC 3.1.21.5) |
|
Recognition Sequence: Non-palindromic Subunit Structure(Restriction Activity): Both R and M-S required Cofactors and Activators: Mg2+, AdoMet(7), ATP (not hydrolyzed)(8), May require a second unmodified site in opposite orientation, variable distance away(9) Cleavage Site: Cuts approximately 25 bases away from the recognition sequence, may not cut to completion. Example: EcoP15I: CAGCAG(N)25-26/ Example(s): EcoP15I, EcoPI, HinfIII, StyLTI |
1R, M and S refer to restriction, methyltransferase, and substrate specificity domains which may exist as separate subunits (R, M, S) or be combined (R-S, M-S, R-M) in a single polypeptide. In the case of Type II systems, the primary sequence of the restriction endonuclease and methyltransferase specificity domains demonstrate little, if any, homology.
2Although showing a strong preference for Mg2+, other divalent metals may substitute, usually Mn2+ but also Co2+, Fe2+, Ni2+, and Zn2+. However, specificity may be relaxed and cleavage rates significantly decreased.
3AatII (14) and SfiI (15) reported to exist as homotetramers.
4 DpnI is the only Type I, II, or III enzyme known which requires 6-methyladenine in its recognition site of GATC for activity.
5Many isoschizomers exist, which are common Type II.
6Eco57 I has been variously classified as Type IIe (6), Type IIs (8), and the only member to date of a new classification, Type IV (16). AdoMet is considered stimulating, but not required for Eco57I, similar to the Type III enzymes.
7AdoMet is considered stimulating, but not required, for all the Type III enzymes (10).
8ATPase activity has been previously reported as <1% compared to Type I restriction activity and therefore ATP was regarded as a cofactor rather than a substrate. However, more recent evidence with EcoP15I (12) suggests a need to investigate possible ATPase activity of Type III restriction activities more closely.
9In the host protection mechanism for EcoP15I, DNA is hemi-methylated in the fully protected state and freshly replicated DNA is protected by the fact that a second, convergently orientated, and also totally unmodified site is required for cleavage. This host protection mechanism may be true for the other Type III systems as well (EcoPI, HinfIII, and StyLTI [12,13]).
References
- Williams, R.J. (in press) In: Methods in Molecular Biology, The Nucleases, Schein, C.H. ed., Humana Press, Totowa, New Jersey.
- Roberts, R.J. and Halford, S.E. (1993) In: Nucleases, Second Edition Linn, S.M., Lloyd, S.R. and Roberts, R.J., eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- Pingoud, A. and Jeltsch, A. (1997) Recognition and cleavage of DNA by type II restriction endonucleases. Eur. J. Biochem. 246, 1–22.
- Kong, H. (1998) Analyzing the functional organization of a novel restriction modification system, the Bcg I system. J. Mol. Biol. 279, 823.
- Sears, L.E. et al. (1996) Bae I, another unusual Bcg I-like restriction endonuclease. Nucleic Acids Res. 24, 3590–2.
- Reuter, M. et al. (1993) Use of specific oligonucleotide duplexes to stimulate cleavage of refractory DNA sites by restriction endonucleases. Anal. Biochem. 209, 232–7.
- Oller, A.R. et al. (1991) Ability of DNA and spermidine to affect the activity of restriction endonucleases from several bacterial species. Biochemistry. 30, 2543-9.
- Szybalski, W. et al. (1991) Class-IIs restriction enzymes--a review. Gene 100, 13–26.
- Belfort, M. and Roberts, R.J. (1997) Homing endonucleases: Keeping the house in order. Nucleic Acids Res. 25, 3379–88.
- Bickle, T.A. (1993) In: Nucleases, Second Edition, Linn S.M., Lloyd, S.R., and Roberts, R.J. eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Wilson, G.G. and Murray, N.E. (1991) Restriction and modification systems. Annu Rev Genet. 25, 585–627.
- Meisel, A. et al. (1995) Type III restriction endonucleases translocate DNA in a reaction driven by recognition site-specific ATP hydrolysis. EMBO J. 14, 2958–66.
- Kruger, D.H. et al. (1995) The significance of distance and orientation of restriction endonuclease recognition sites in viral DNA genomes. FEMS Microbiol. Rev. 17, 177–84.
Restriction Enzyme Structure and Mechanism of Action
There is little amino acid sequence homology between the nuclease and methylase within a restriction/modification system, even among the regions responsible for recognition. Among restriction enzymes, exact isoschizomers isolated from bacteria of the same genus can show little or no similarity in their methylation sensitivity, digestion optima or primary sequence except for a limited PD…D/EXK motif involved in catalysis. However, this common motif has been found in Type II,IIb, IIe, IIs and in intron-encoded restriction enzymes (1) (2).
Despite the lack of primary sequence homology, three-dimensional structure among Type II homodimers is similar for those enzymes where crystallography data is available. In general, the holoenzyme dimer resembles a "U" shape, with each side constituting a monomer containing both recognition and catalytic domains with an overlapping bridging domain at the bottom. The DNA is bound between the two subunits. FokI, the most studied Type IIs enzyme, appears to exist primarily as a monomer but transiently forms a similar dimer at the recognition site (3).
Restriction endonucleases bind dsDNA both specifically and non-specifically. After binding at a non-cognate sequence, several enzymes have been shown to locate their targets through linear diffusion. For example, EcoRI diffuses along linear DNA at a rate of approximately 7 x 106bp s-1 (4) and EcoRV diffuses at approximately 1.7 x 106bp s-1 (5). During this process a large number of water molecules appear to fill the spaces between the enzyme and the DNA. Once the cognate (recognition) sequence is found, much of the water is excluded as a highly redundant number of contacts evolve between the enzyme and the bases and phosphodiester backbone of the DNA. In the case of EcoRI, 50 water molecules are excluded at the cognate site (6). Generally, 2-3 non-specific bases on either side of the target sequence are required for proper recognition. Conformational changes occur in both the enzyme and DNA as the specific complex forms. The resulting induced fit positions the catalytic center in reactive proximity to the substrate. For most enzymes studied to date, this is able to occur in the absence of Mg2+.
Using the known co-crystal structures of enzymes bound to their cognate sequences and substitution experiments in the enzyme or DNA for a limited number of additional enzymes, a mechanism for DNA cleavage has been postulated. Evidence for most enzymes studied to date supports a substrate assisted catalysis model (7). In this model, conserved amino acids at the catalytic site bind Mg2+ and position it near the scissile phosphate. Hydrolysis begins by in-line nucleophilic attack of an activated water molecule. The phosphate 3´ of the scissile phosphorous has been shown to play some role in catalysis, most likely in activating the water, as greatly reduced cleavage occurs when a methylphosphonate (8) or phosphothioate (9) occupy this position. A conserved lysine and/or a Mg2+ also may be involved in activating the water and stabilizing the pentavalent transition state produced at the scissile phosphorous (10). Inversion occurs as the 3´-OH leaving group is protonated by a Mg2+-bound water upon exit.
Regardless of the mechanism of action, all restriction enzymes share two common features, a requirement for Mg2+, and 5´-phosphate and 3´-OH products. Some enzymes may also need AdoMet or ATP, and/or binding of a second recognition sequence to an allosteric site on the enzyme as a requirement for, or a stimulator of, cleavage.
References
- Wilson, G.G. and Murray, N.E. (1991) Restriction and modification systems. Annu Rev Genet. 25, 585–627.
- Stahl, F. et al. (1998) The mechanism of DNA cleavage by the type II restriction enzyme EcoR V: Asp36 is not directly involved in DNA cleavage but serves to couple indirect readout to catalysis. Biol. Chem. 379, 467–73.
- Bitinaite, J. et al. (1998) Fok I dimerization is required for DNA cleavage. Proc Natl Acad Sci U S A. 95, 10570.
- Ehbrecht, H.J. et al. (1985) Linear diffusion of restriction endonucleases on DNA. J. Biol. Chem. 260, 6160–6.
- Jeltsch, A. and Pingoud, A. (1998) Kinetic characterization of linear diffusion of the restriction endonuclease EcoR V on DNA. Biochem. 37, 2160–9.
- Robinson, C.R. and Sligar, S.G. (1998) Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proc. Natl. Acad. Sci. USA 95, 2186–91.
- Pingoud, A. and Jeltsch, A. (1997) Recognition and cleavage of DNA by type II restriction endonucleases. Eur. J. Biochem. 246, 1–22.
- Jeltsch, A. et al. (1995) Evidence for substrate-assisted catalysis in the DNA cleavage of several restriction endonucleases. Gene. 157, 157–62.
- Jeltsch, A. et al. (1993) Substrate-assisted catalysis in the cleavage of DNA by the EcoR I and EcoR V restriction enzymes Proc Natl Acad Sci U S A. 90, 8499–503.
- Sam, M.D. and Perona, J.J. (1999) Catalytic roles of divalent, metal ions in phosphoryl transfer by EcoR V endonuclease. Biochemistry 38, 6576–86.
Star Activity
The precise specificity of the approximately 3,000 known restriction enzymes for their >200 different target sequences could be considered their most interesting characteristic. Although all restriction enzymes bind DNA nonspecifically, under optimal conditions the difference in cleavage rates at the cognate site and the next best site (single base substitution) is very high. For example, the rate difference for EcoRI at its cognate site (5´-GAATTC-3´) and next best site (5´-TAATTC-3´) is of the order of 105 (1) . Similarly, for EcoRV, cleavage at its cognate site (5´-GATATC-3´) is 106 times faster than at the next best site (5´-GTTATC-3´) (2) .
However, under non-optimal conditions, the differences in cleavage rates between cognate and next-best sites change dramatically for many enzymes. This loss of fidelity or increase in cleavage at sites similar to the cognate site is commonly referred to as star activity. A number of reaction parameters can increase the rate of cleavage at star sites relative to cognate sites. These include pH, type of ions present, ionic strength, metal cofactors other than Mg2+, high DNA:enzyme ratios and the presence of volume excluders (glycerol, ethylene glycol, etc.). In conjunction with this increase in star activity, cleavage rates at the cognate site generally decrease. For example, for EcoRI, the rate difference between cognate and star sites approaches zero as ethylene glycol concentration increases up to 4M (3) and for EcoRV, the rate difference drops to only 6-fold when Mn2+ is substituted for Mg2+ (2) .
Several plausible explanations for star activity are based on the proposed mechanisms for target site identification and hydrolysis (see Structure and Mechanism of Action for more information). During nonspecific binding, a large number of water molecules are present at the protein-DNA interface. When tighter binding and positioning of the catalytic site occurs upon recognition of the target sequence, the number of these interface water molecules is significantly reduced. The higher osmotic pressure caused by volume excluders results in the same reduction in the amount of interface water molecules and allows easier active complex formation at star sites (3) . At alkaline pH, higher OH- concentrations may reduce the need for an activated water molecule, which normally initiates nucleophilic attack on the scissile phosphorous. Mn2+ has a higher affinity for oxygen ligands than Mg2+and may bind more easily to a catalytic site in a partially active conformation at a star site. Also, it is possible that Mn2+-bound water is better able to protonate the leaving group since it has a lower pKa than Mg2+ bound water (4) .
Although all restriction enzymes probably exhibit some decrease in the cleavage rate difference between cognate and near-cognate sites under such extreme conditions as 4M ethylene glycol, most are not significantly affected under common usage conditions. Those that are susceptible to star activity are induced to different degrees by variations in reaction conditions or by combinations of the conditions listed above. The Table below lists the enzymes sold by Promega that may exhibit star activity, especially under reaction conditions that deviate from those recommended. In multiple enzyme digests or multiple step applications, it is advisable to stay at or near the optimal conditions for these enzymes whenever possible.
Promega Enzymes That May Exhibit Star Activity.
| BamHI | HindIII |
PstI |
SgfI |
| BclI | KpnI |
SalI |
|
| EcoRI | NdeI | SacI |
References
- Lesser, D.R., Kurpiewski, M.R. and Jen-Jacobson, L. (1990) The energetic basis of specificity in the EcoRI endonuclease--DNA interaction. Science 250, 776–86.
- Vermote, C.L. and Halford, S.E. (1992) EcoR V restriction endonuclease: Communication between catalytic metal ions and DNA recognition. Biochemistry 31, 6082–9.
- Robinson, C.R. and Sligar, S.G. (1998) Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proc. Natl. Acad. Sci. USA 95, 2186–91.
- Pingoud, A. and Jeltsch, A. (1997) Recognition and cleavage of DNA by type II restriction endonucleases. Eur. J. Biochem. 246, 1–22.
Site Preferences
When presented with multiple recognition sites that differ in their flanking sequences, most restriction enzymes exhibit slight preferences and cleave the sites at different rates. These rate differences are such that the addition of a small excess of enzyme will avoid any problems due to incomplete digestion. As always, however, one must be aware of the experimental molar concentration of recognition sites and digest conditions relative to that of the unit definition. See Substrate Considerations for further information.
A. Type IIe Restriction Enzymes
A few restriction enzymes have considerably greater difficulty in cleaving some of their recognition sites. Original experiments with these enzymes led to designation of their site preferences as shown:
Restriction Enzyme Site Preferences |
|
| cleavable sites | >90% cleavage with 1-5 fold excess enzyme |
| slow sites | 5-90% cleavage with 1-5 fold excess and additional cleavage with 10-30 fold excess |
| resistant sites | <5% cleavage with 5 fold excess enzyme and no additional cleavage with 10-30 fold excess |
Enzymes that have cleavable, slow, and resistant sites in the same or different DNAs have been designated Type IIe restriction enzymes. This group is comprised of enzymes that would otherwise be members of the common Type II or Type IIs classes. The Type IIe enzymes are NaeI, NarI, BspMI, HpaII, SacII, EcoRII, AtuBI, Crf9I, SauBMKI, and Ksp632I (1) . There is evidence to suggest that Eco57I also belongs to this group (2) .
B. Effector Sequences
Investigation revealed that binding of a second recognition sequence, in cis or trans, to a distal, non-catalytic site on the enzyme allows slow and resistant sites to become cleavable. This effector sequence alters the kinetics in one of two ways. In the K class (NarI, HpaII, SacII), activator DNA binding decreases the Km without altering the Vmax of cleavage, indicating that cooperative binding induces a conformational shift that increases the affinity of the enzyme for its substrate. In the V class (NaeI, BspMI), binding of activator DNA increases the Vmax without changing the Km, indicating that the increased catalytic activity is not related to the affinity of the enzyme for its substrate . It is assumed that the flanking sequences of a recognition site influence the kinetics of cleavage at that site, but at this time the interaction is not understood. Considerable differences also exist in the ability of effector sequences to stimulate cleavage. Generally, a recognition site flanked by the sequence from a site that is cleaved easily is a useful starting point for designing good effector sequences.
References
- Oller, A.R. et al. (1991) Ability of DNA and spermidine to affect the activity of restriction endonucleases from several bacterial species. Biochemistry. 30, 2543-9.
- Reuter, M. et al. (1993) Use of specific oligonucleotide duplexes to stimulate cleavage of refractory DNA sites by restriction endonucleases. Anal. Biochem. 209, 232–7.
Standard Restriction Enzyme Reactions
Each restriction enzyme has optimal reaction (assay) conditions and different conditions for long term storage. The recommended assay and storage conditions are both determined by the manufacturer to provide the user with the highest activity, best fidelity and greatest stability for each enzyme. Factors that must be considered include temperature, pH, enzyme cofactors, salt composition, ionic strength and stabilizers. Promega restriction enzyme Reaction Buffers are designed to provide the best balance of optimal activity and convenience. Promega storage buffers have been designed after accelerated and real time/real temperature stability experiments. All enzyme storage conditions are validated through our Quality Assurance re-assay program to maximize long term stability.
Setting up digests with a single restriction enzyme is relatively straightforward. However, digests using multiple enzymes that have different buffer requirements may demand the use of alternative buffers and may require adjustments in the number of units of enzyme used. See the Reference Table Relative Activity of Restriction Enzymes in Promega 10X Buffers or use the Restriction Enzymes interactive search tool. If no compatible buffer can be found a sequential reaction may be performed in which additional buffer or salt is added to the reaction before the second enzyme, or each digest may be performed sequentially using the optimal buffers. The latter option will require either a DNA precipitation or purification step after the first digest. Regardless of the type of digest performed, the addition of BSA is recommended to stabilize the enzyme and enhance activity (1)(2).
A. Reaction Conditions
pH: Most restriction enzymes are used between pH 7.2 and pH 8.5 as measured at the temperature of incubation. pH values outside of the optimal range may lead to star activity.
Mg2+: Commercially available restriction enzymes require Mg2+ as the only cofactor. Restriction enzyme activities are relatively insensitive to the Mg2+ concentration; similar rates are observed from 5-30mM. The presence of other divalent metal ions, especially Mn2+, may lead to star activity.
Salt Concentration: Restriction enzymes are diverse in their response to ionic strength. Most are stimulated by 50-150mM NaCl or KCl while others are inhibited by salt concentrations higher than 20mM. A few enzymes prefer acetate to chloride anions. Suboptimal ionic strength or type of ion may lead to star activity.
BSA: Bovine Serum Albumin is used in restriction enzyme storage buffers and is added to digestion reactions to stabilize the enzyme. BSA can protect restriction enzymes from proteases, non- specific adsorption and harmful environmental factors such as heat, surface tension and interfering substances. Typically, the addition of 0.1mg/ml BSA will result in a 1.5 to 6-fold enhancement of enzyme activity. The Acetylated BSA provided with Promega's restriction enzymes has been modified and extensively tested to ensure that no degrading activities are present.
Glycerol: Glycerol is added to restriction enzyme storage buffers to prevent freezing at -20°C. Repeated freeze/thawing of restriction enzymes can reduce their activity. Some restriction enzymes show reduced specificity, or increased star activity, when the glycerol concentration in the final reaction is higher than 5% although many have normal specificity at glycerol concentrations as high as 10%.
Incubation Temperature: Most restriction enzymes show maximum activity at 37°C. A few enzymes require higher or lower temperatures for optimal activity (e.g., TaqI, 65°C; SmaI, 25°C). For incubations greater than 1 hour with high temperature enzymes, cover the reactions with a drop of mineral oil to prevent evaporation. Generally, the incubation temperature for the enzyme reflects the growth temperature of the bacterial strain from which it is derived. For enzymes that have temperature optima other than 37°C, Promega provides information on percent activity at 37°C on the Product Information sheet that is packaged with each enzyme. This type of information is particularly useful when performing double digests.
Volume: Viscous DNA solutions inhibit enzyme diffusion and can reduce enzyme activity. DNA concentrations that are too dilute can fall below the Km of the restriction enzyme and also affect enzyme activity. Volume considerations must take into account final ionic strength and must result in glycerol concentrations no higher than 5-10% in order to avoid star activity. Reaction volumes of 10-50µl per microgram of DNA are recommended.
B. Single Restriction Enzyme Digests
An analytical restriction enzyme reaction is usually performed in a volume of approximately 20µl on 0.2-1.5µg of substrate DNA using a 2- to 10-fold excess of enzyme over DNA, based on unit definition. Use of an unusually large volume of DNA or enzyme may give aberrant results. Caution should be exercised to prevent higher than normal concentrations of EDTA and glycerol. The following is an example of a typical analytical single restriction enzyme digestion:
- Under sterile conditions add the following components, in the order stated, to a sterile microcentrifuge tube.
Sterile, nuclease-free water 14µl Restriction enzyme 10X buffer 2µl BSA, Acetylated (1mg/ml) 2µl DNA sample 0.2-1µg, in water or TE buffer 1µl Restriction enzyme, 2-10U 1µl Final volume 20µl - Mix gently by pipetting. Centrifuge briefly at 12,000 x g in a micro centrifuge to collect the contents at the bottom of the tube.
- Incubate at the optimum temperature for 1-4 hours.
- Add 4µl of Blue/Orange 6X Loading Dye (or another appropriate DNA loading buffer), and proceed to gel analysis.
Larger scale restriction enzyme digestions can be accomplished by scaling this basic reaction proportionately.
C. Multiple Restriction Enzyme Digests
If all of the restriction enzymes in a multiple digest have the same optimal buffer, setting up the digest is straightforward. However, when this is not the case, several options are available.
- Use the optimal buffer supplied with one enzyme if the activity of the second enzyme is acceptable in that same buffer. Alternatively, acceptable activity for both enzymes may be achieved by using another of Promega’s 4-CORE® 10X Buffers(Cat.# R9921). If one of the enzymes has less than 75% activity in the chosen buffer, the reaction time or the number of units of enzyme used may need to be increased. Be aware of possible star activity under non-optimal reaction conditions (See the Reference Table Relative Activity of Restriction Enzymes in Promega 10X Buffers or use the Restriction Enzymes interactive search tool to identify compatible buffers).
- Choose an isoschizomer or neoschizomer with more compatible buffer requirements.
- Perform a single digest with the first enzyme then inactivate that enzyme. Add the ingredients necessary for the second digest then add the second enzyme. For example, use a lower salt buffer and enzyme first, then inactivate the first enzyme, add enough salt to achieve the concentration required for the second digest, and add the second restriction enzyme.
Note: Perform each digest sequentially using the optimal buffers. This will require either a DNA precipitation or purification step after the first digest. Although this procedure involves more steps than those listed above, in situations where options 1-3 are not satisfactory, it may be the best alternative.
D. Experimental Controls
Some common controls used for restriction enzyme digestion and gel analysis are given in the Table below.
Restriction Enzyme Reaction Controls |
|
| Restriction Enzyme Digest Controls | |
| Control: Untreated DNA control | |
| Strategy | Purpose |
| DNA is loaded on gel with no treatment other than the addition of loading buffer. | Shows the integrity of the DNA starting material. Nicked, linear and supercoiled forms of plasmid DNA are normally seen in untreated samples. |
| Control: No enzyme Control | |
| Strategy | Purpose |
| A mock digest is run parallel with the experimental digest, except that no enzyme is added. The missing volume is made up with water. | Compares DNA digests with and without enzyme. Detects changes that may occur independent of enzyme such as exonuclease contamination in the DNA or in one of the reaction components. |
| Control: Enzyme activity check | |
| Strategy | Purpose |
| Perform a control digest using the unit definition DNA (usually lambda) and conditions as described in the Promega Product Information sheet. | Confirms enzyme activity. |
| Control: DNA substrate control and general enzyme digest control | |
| Strategy | Purpose |
Set up the following parallel digests:
|
Compares activity of the enzyme under experimental conditions using standard DNA and experimental DNA under standard conditions. Tests for possible problems with substrate DNA such as impurity, missing recognition sites, methylation, etc. Can be used to assay for the function of other reagents used in the enzyme digest. If an inhibitor is suspected in the DNA solution, a set of digests comparing experimental DNA, control DNA and a combination of the two may also be performed. In most cases, the presence of an inhibitor will "poison" the control reaction when both are combined. |
| Gel Analysis Controls | |
| Control: One molecular weight (MW) marker | |
| Strategy | Purpose |
| One or two lanes of an electrophoresis gel should always be devoted to size standards and used for comparison with unknowns. | This assures that a standard exists for:
|
| Control: Two different MW markers | |
| Strategy | Purpose |
| Two different size markers provide much more information than one. Two sets of data points give greater accuracy during graphing of data points for MW measurements (by comparison with the mobility of the standards). Lane-to-lane variation may also be detected if two standards are used but they migrate differently. | A greater range of size standards permits more accurate size estimation, and allows identification of conformational effects on mobility as well as electrophoretic variability. Anomalous mobility due to differences in the quantity of sample loaded may also be detected. |
| Control: Load two different quantities of the same MW marker on the gel | |
| Strategy | Purpose |
| Mass per band control: Loading two different quantities of the same size marker will yield important information about mobility shifts due to mass per band differences. | Allows detection of mass effects on mobility. Also may show lane-to-lane variation during gel electrophoresis. |
| Control: Salt effects control | |
| Strategy | Purpose |
| Run markers beside unknown with and without the salt contained in the experimental digest. | Detects any gel retardation that may occur due to the presence of high salt concentrations in sample. |
| Control: Markers of known mass are run in a dilution series | |
| Strategy | Purpose |
| Bands of similar MW are chosen in marker and experimental lanes. The mass of the band in question is compared to a control based on its staining intensity. It is crucial that many dilutions are run side-by-side in order to achieve the most accurate visual comparison. | The quantity of an unknown DNA sample may be assessed in this manner or used to confirm a result obtained by spectrophotometry. |
References
- Williams, R., Kline, M. and Smith, R. (1996) BSA and restriction enzyme digestions. Promega Notes 59, 46.
- Lepinske, M. (1996) BSA and restriction enzyme digestions. Promega Notes 60, 28.
Restriction Enzyme Activity
Restriction enzymes differ in their reaction kinetics. As a result, variations from the recommended incubation time, number of units used, substrate amount, and/or total reaction volume should be considered carefully to ensure complete digestion. The following table gives an indication of the activity of Promega blue/white cloning-qualified and genome-qualified restriction enzymes under varying reaction conditions. Variations in the number of enzyme units used and the reaction incubation times were tested. In each case the reaction volume (50µl) and the amount/type of DNA substrate (1µg) were the same as that used in the unit definition assay. Incubation time for the unit definition assay is one hour.
Restriction Enzyme Activity under Nonstandard Units and Incubation Time Conditions |
|||||||
| Enzyme | Reaction Time and Number of Units Used | ||||||
| 15
min. 4 units |
15
min. 2 units |
15
min. 1 unit |
30
min. 2 units |
1
hr 1 unit |
2
hr 0.5 units |
4
hr 0.25 units |
|
| ApaI | C | I | I | C | C | C | C |
| BamHI | C | C | I | C | C | I | I |
| BclI | C | I | I | C | C | C | C |
| BglI | C | I | I | C | C | C | C |
| ClaI | C | I | I | C | C | C | C |
| EcoRI | C | I | I | C | C | C | C |
| EcoRV | C | I | I | C | C | C | C |
| HindIII | C | I | I | I | C | C | C |
| KpnI | C | I | I | C | C | C | C |
| MluI | C | I | I | C | C | C | C |
| NcoI | C | I | I | C | C | C | C |
| NheI | C | I | I | C | C | C | C |
| NotI | C | I | I | C | C | C | I |
| PstI | C | I | I | C | C | C | C |
| SacI | C | I | I | C | C | C | C |
| SacII | C | I | I | C | C | C | C |
| SalI | C | C | C | C | C | C | I |
| SmaI | C | C | I | C | C | C | C |
| SpeI | C | I | I | C | C | I | I |
| SphI | C | C | C | C | C | I | I |
| XbaI | C | I | I | C | C | C | C |
| XhoI | C | I | I | C | C | C | C |
"C" indicates complete cleavage; "I" indicates incomplete cleavage for the units and incubation times shown.
Back to topRestriction Enzyme Substrate Considerations
A. Substrate Source and Structure
Substrates commonly used for restriction enzyme digestion include phage DNA, plasmid DNA, genomic DNA, PCR products and double-stranded oligonucleotides. The concentration of the DNA sample can influence the success of a restriction digestion. Viscous DNA solutions, resulting from large amounts of DNA in too small of a volume, can inhibit diffusion and can significantly reduce enzyme activity (1). DNA concentrations that are too low also may inhibit enzyme activity (see Substrate Quality). Typical Km values for restriction enzymes are between 1nM and 10nM, and are template-dependent (2). Recommended final DNA concentrations for digestion range from 0.02-0.2µg/µl. Substrate structural variations, concentration and special considerations are discussed below according to DNA type.
Lambda DNA: Lambda DNA is a linear DNA that is an industry standard for the measurement and expression of unit activity for most restriction enzymes. In general, one unit is sufficient to cut 1µg of lambda DNA in 1 hour under optimal reaction conditions in a reaction volume of 50µl. In lambda DNA, the cos ends, (12-base, complementary, single-stranded overhangs at the end of each molecule) may re-anneal during digestion. This can give the appearance that digestion is incomplete. To avoid this problem, heat the DNA at 65°C for 5 minutes prior to electrophoresis to melt ends that have annealed.
Plasmid DNA: Circular, supercoiled plasmid DNA typically ranges from 3-10kb in size. Compared to linear DNA, plasmids often require more units of restriction enzyme for complete cleavage due to the supercoiling(1) or the total number of sites to be digested (see Recognition Site Density). See Digestion of Supercoiled Plasmid DNA for information on the relative units needed for complete cleavage of a typical plasmid vector with common cloning enzymes. If a supercoiled plasmid is first linearized with another restriction enzyme or relaxed with topoisomerase, less enzyme may be needed for digestion.
Genomic DNA: Digestion of genomic DNA can be difficult due to methylation and viscosity. If methylation is a concern, consider using isoschizomers with different methylation sensitivities (see Methylation Sensitivity of Isoschizomer/Neoschizomer Pairs). Viscosity can be adjusted by increasing the reaction volume. Genomic DNA often digests more efficiently when it is diluted to a minimum concentration of 10µg per 50-200µl. If this is not possible, heating the DNA at 65ºC for ten minutes prior to the addition of the restriction enzyme can enhance activity (3). Addition of spermidine to final concentration of 1-5mM also has been reported to increase enzyme activity in the digestion of genomic DNA (4). Addition of BSA to restriction digests at a final concentration of 0.1mg/ml may also improve enzyme activity.
PCR Products: PCR-amplified DNA may be digested with restriction enzymes that have recognition sequences within the amplified sequence or in the primer regions. The number of enzyme units needed must be balanced with the total number of sites to assure complete cleavage. Longer incubation times may be required to ensure complete digestion. Enzymes with low overdigestion values (<12 units/16 hours) should be avoided in overnight digestions, as star activity or trace contaminants present in these enzymes may lead to problems. Consult the Promega Product Information sheet for the overdigestion value of the enzyme. For many common restriction enzymes, acceptable activity is seen in PCR buffer, although digestion after amplification may not result in the expected compatible ends due to residual polymerase activity (5). Digestion near the end of a PCR product may also present problems. Restriction enzymes require varying amounts of flanking DNA around the recognition site, usually 1-3 bases but occasionally more (See Digestion of Sites Close to the End of Linear DNA). If an oligonucleotide primer is designed with a cut site that is too close to the end of the DNA, the site may cut poorly or not at all. Since it is very difficult to assay for cutting near the end of DNA, the effectiveness of compensation with extra enzyme units or increased incubation time is difficult to determine. Use of proofreading enzymes in PCR may also complicate the situation as these enzymes are capable of degrading the 3´ ends of amplimers, interfering with complete digestion by restriction enzymes. The use of high dNTP concentrations and immediate cooling to 4°C after PCR will reduce such degradation. Another reason for incomplete digestion of PCR fragments may be primer dimers. If the restriction site is built into the primer, primer dimers will contain a double-stranded version of the site, usually in vast molar excess over that of the desired target PCR fragment. This problem can be easily avoided by purifying the PCR fragment prior to restriction enzyme digestion using the Wizard® PCR Preps DNA Purification System (Cat.# A7170).
Double-Stranded Oligonucleotides: Many of the same considerations for PCR products apply to the digestion of double-stranded oligonucleotides. In this case high densities of recognition sites per unit of mass can be present and the site may also be near the end of the DNA molecule. Again, longer digestion times and/or more enzyme may be needed. Enzymes with a low overdigestion specification (12 units/16 hours) should be avoided in overnight digestions.
Single-Stranded DNA: Cleavage of single-stranded DNA, although at a greatly reduced rate compared with double-stranded DNA, has been reported for a few restriction enzymes (6). Studies have shown, however, that several restriction enzymes that appear to cleave single-stranded DNA actually recognize folded-back duplex regions within the single-stranded genomes (e.g., M13, f1, single-stranded phiX174) (7) (8). Therefore, these enzymes are not digesting single-stranded DNA, rather individual sites that are in the duplex form.
DNA-RNA Hybrids: Digestion of DNA-RNA hybrid molecules has been described for several restriction enzymes (AluI, EcoRI, HaeIII, HhaI, HindIII, MspI, SalI, ThaI) (9). In these cases, the DNA strand of the hybrid was digested in the identical place as duplex DNA. Digestion required 20 to 50-fold higher enzyme levels than those needed for duplex DNA. It is possible but not proven that the RNA was also cleaved with large excesses of enzyme.
Influence of Flanking Sequence: The sequences flanking the restriction enzyme recognition sequence can influence the cleavage rate of many restriction enzymes although the differences are usually less than 10-fold. A small number of enzymes (e.g., NaeI, HpaII, SacII, NarI, EcoRII) exhibit more pronounced site preferences and are designated Type IIe. See Site Preferences and Turbo™ Restriction Enzymes for further information.
Methylation: Methylation of nucleotides within restriction enzyme recognition sequences can affect digestion. Methylation may occur as 4-methylcytosine, 5-methylcytosine, 5-hydroxymethylcytosine or 6-methyladenine in DNA from bacteria (including plasmids), eukaryotes and their viruses. The sensitivity, or lack thereof, to site-specific methylation, is known for many restriction enzymes (10). Often, isoschizomers differ in their methylation sensitivity. Refer to Cat.# A1330) provide an easy and effective way to isolate and purify DNA, free of salt or macromolecular contaminants. The addition of spermidine to a final concentration of 1mM and/or BSA to a final concentration of 0.1mg/ml can also improve digestion of poor quality miniprep DNA.
Genomic DNA: Genomic DNA frequently contains more contaminants than plasmid DNA. Best results are obtained when the absorbance ratios at A260/A280 are at least 1.8. Spermidine can be added to a final concentration of 1mM and/or BSA to a final concentration of 0.1mg/ml to improve digestion of poor quality genomic DNA. For further information see Digestion of High Molecular Weight DNA.
Genomic DNA Embedded in Agarose plugs: Pulsed field gel electrophoresis permits the resolution of extremely large DNA fragments. Genomic DNA purified by traditional techniques can contain double-stranded breaks due to mechanical shear forces. Such breaks can be a source of background in megabase mapping of fragments of 50-1000kb. To avoid this, mammalian, bacterial and yeast cells can be embedded in agarose strips and the cells lysed and treated with proteinase K in situ (11). Most restriction enzymes can cut DNA embedded in agarose provided that more enzyme and longer incubation times are used. A good rule of thumb is to use 5-10 units of enzyme per microgram of DNA and to avoid using restriction enzymes with low overdigestion values (<20 units/16 hours), which can cause problems during longer incubations with excess enzyme. For further information, refer to Digestion of High Molecular Weight DNA.
Genomic DNA Purified From Blood. The anti-coagulant used during blood collection can affect the ability of restriction enzymes to completely digest DNA. Use EDTA as an anti-coagulant rather than Heparin, which can bind tightly to the enzyme and interfere with digestion. The absorbance ratios at A260/A280 should be at least 1.8, indicating that protein has been removed efficiently. A number of rapid DNA purification protocols have been written that do not require separation of white cells from red cells (12) (13). These techniques can yield good quality DNA from small volumes of blood, but the DNA obtained after scale-up may be of poorer quality. For larger blood samples, a technique that separates white blood cells from red blood cells, such as pelleting red blood cells through a Ficoll® gradient, is recommended prior to DNA purification.
Promega offers the Wizard® Genomic DNA Purification Kit (Cat.# A1120) for the isolation of genomic DNA from white blood cells (with reagents/protocol for removal of red cells), tissue cultured cells, animal tissue, plant tissue and Gram-positive and Gram-negative bacteria. DNA purified with this system is suitable for digestion with restriction enzymes.
PCR Products: Contaminants in PCR such as salts, glycerol, and primer dimers can inhibit restriction enzyme activity. The Wizard® PCR Preps DNA Purification System (Cat.# A7170) provides a reliable method for purification of double-stranded PCR-amplified DNA from any salts or macromolecular contaminants.
C. Recognition Site Density
Restriction enzyme activity units are usually defined based on a one-hour digest of 1µg of lambda DNA. When digesting other substrates, adjustments may be needed based on the amount of substrate, the number of recognition sites per molecule and the incubation time. The following table illustrates the effect of differences in substrate recognition sites per molecule for EcoRI while keeping the substrate mass and incubation time constant.
Differences in Substrate Recognition Sites for EcoRI |
|||||
| DNA Substrate | Base Pairs |
Picomoles in 1µg* |
Cut Sites (EcoRI) |
Picomoles Cut Sites |
Units Needed |
| Unit definition (lambda) | 48,502 | 0.0317 | 5 | 0.1585 | 1 |
| plasmid | 3,000 | 0.5 | 1 | 0.5 | 3** |
| PCR fragment | 700 | 2.2 | 1 | 2.2 | 14 |
| oligonucleotide | 25 | 62.5 | 1 | 62.5 | 394 |
*Based on 650 Daltons per base pair of DNA.
**Enzymes differ in their ability to digest supercoiled vs. linear substrates.
References
- Fuchs, R. and Blakesley, R. (1983) Guide to the use of type II restriction endonucleases. Meth. Enzymol. 101, 3.
- Wells, R., Klein, R. and Singleton, C.K. (1981) In: The Enzymes XIV 157.
- Hinds, K., Shamblott, M. and Litman, G. (1991) In: Methods in Nucleic Acid Research, Karam, J., Chao, L. and Warr, G. eds., CRC Press.
- Bloch, K. (1987) In: Current Protocols in Molecular Biology, Ausubel, F.M. et al., eds., Green Publishing Associates.
- Turbett, G.V. and Sellner, L.N. (1996) Digestion of PCR and RT-PCR products With restriction endonucleases without prior purification or precipitation. Promega Notes 60, 23–7.
- Yoo, O.J. and Agarwal, K. L. (1980) Cleavage of single strand oligonucleotides and bacteriophage phiX174 DNA by Msp I endonuclease. J. Biol. Chem. 255, 10559–62.
- Nevendorf, S. and Wells, R. (1980) In: Gene Amplification and Analysis: Restriction Endonucleases. Vol. I, Chirikjian, J., ed., Elsevier, North Holland.
- Blakesley, R.W. et al. (1977) Duplex regions in "single-stranded" phiX174 DNA are cleaved by a restriction endonuclease from Haemophilus aegyptius. J. Biol. Chem. 252, 7300–6.
- Molloy, P.L. and Symons, R.H. (1980) Cleavage of DNA.RNA hybrids by type II restriction enzymes. Nucleic Acids Res. 8, 2939.
- McClelland, M. et al. (1994) Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 22, 3640–59.
- McClelland, M. et al. (1987) Restriction endonucleases for pulsed field mapping of bacterial genomes. Nucleic Acids Res. 15, 5985–6005.
- Miller, S.A., Dykes, D.D. and Polesky H.F. (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215.
- Grimberg, J. et al. (1989) A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 17, 8390.
Digestion of High Molecular Weight DNA
A. Isolation of High Molecular Weight DNA
High molecular weight genomic DNA can be prepared using several different methods including traditional phenol extraction (1), standard isolation procedures such as the Wizard® Genomic DNA Purification Kit (Cat.# A1120) (2) (3) or by embedding the cells of interest in blocks or beads of agarose and enzymatically digesting the cell membranes and proteins (4). Large DNA is quite susceptible to mechanical shearing and it is difficult to obtain DNA of 50kb or more unless it is embedded in agarose. Regardless of the preparation method, genomic DNA is frequently less pure than plasmid or other smaller DNA that can be treated more harshly during isolation. In addition, genomic DNA, especially that of higher organisms, may contain more modifications such as methylation. The methylation sensitivity of potential restriction enzymes may need to be considered for genomic digests. Excess restriction enzyme units and extended incubation times are standard for genomic digestions. For long incubations, especially at elevated temperatures, evaporation of water from the buffer can concentrate components of the reaction and cause star activity. The reaction can be overlaid with mineral oil or the digestion performed in an incubator to avoid evaporation. Addition of spermidine to a final concentration of 1-5mM has also been shown to be helpful for genomic digests (1) (5).
For DNA in solution, such as that prepared by phenol extraction or by using the Wizard® Genomic DNA Purification Kit, viscosity may limit mixing of solutions and diffusion of the enzyme. In general 2-10 units of restriction enzyme per microgram of DNA in a reaction volume of 20µl is recommended. Incubation time is typically 4 hours to overnight.
A general protocol for embedding and digesting mammalian cells in agarose is provided below. Conditions will differ significantly for other cell types.
B. Procedure for Embedding Mammalian Cells in Agarose Gel Plugs
- Harvest the cells by centrifugation at 500 x g for 10 minutes. Wash the cells in an isotonic solution and resuspend in the same solution. Count the cells and dilute them to an appropriate density (~5 x 107 cells/ml) in isotonic buffer.
- Make 1% low melting temperature agarose in isotonic buffer, heat to melt the agarose then cool to 37°C.
- Warm the cell suspension to 37°C, mix 1:1 with the melted agarose and dispense into appropriate molds. It is best to keep the molds on ice so that the agarose will gel rapidly. This will reduce settling of the cells.
- After the plugs have set, remove them from the mold and place them in a solution of 1mg/ml pronase in 1% lauryl sarcosine, 0.5M EDTA, and 10mM Tris (pH 9.5).
- Leave the plugs in this solution at room temperature for 30 minutes to allow diffusion of the pronase and buffer into the plugs.
- Incubate at 50°C overnight. Replace the buffer/pronase, and incubate at 50°C for another 24 hours.
- Rinse the plugs in buffer for 2 hours, repeat this rinse once more. Store at 4°C.
C. Digestion of High Molecular Weight DNA Embedded in Agarose Gel Plugs
The conditions required for digestion of agarose-embedded DNA differ from those required for digestion of DNA in solution. In general, much more restriction enzyme is needed. We have tested a number of enzymes for their ability to digest DNA embedded in agarose (see Table below). The exact amount of enzyme needed varies depending on the DNA type and preparation. A general protocol for digestion of agarose embedded DNA is provided below.
Protocol
- Soak the agarose plug in TE buffer (10mM Tris-HCl [pH 7.4], 1mM EDTA) for 30 minutes on ice.
- Equilibrate the plug in the appropriate restriction enzyme buffer supplemented with 20µg/ml of BSA for 30 minutes on ice.
- Add the restriction enzyme to each tube (see Table below for examples of the appropriate amount of enzyme to use). Allow the enzyme to diffuse into the agarose for 30 minutes on ice.
- Incubate the reaction at the appropriate temperature for 3 hours to overnight.
- Add EDTA to a final concentration of 60mM to stop the reaction.
- The digested agarose plug can be stored at 4°C for several days until use.
Parameters for Digestion of Chromosomal DNA by Promega Genome-Qualified Restriction Enzymes |
||||||||
| Genome | Conditions for Digestion | |||||||
| Promega Enzyme |
Recognition Sequence |
Source | Size (Mb) |
G+C (%) |
Enzyme (u): DNA (µg) |
Temp. (°C) |
Time (hr) |
Number of Fragments |
| BclI | T/GATCA | N. crassa | 45 | 54 | 30:2 | 50* | 15 | many |
| BglI | GCCNNNN/NGGC | S. aureus | 3.0 | 34 | 30:2 | 37 | 3 | 20-25 |
| ClaI | AT/CGAT | M. bovis | 2.9 | 45 | 12:1 | 37 | 4 | many |
| MluI | A/CGCGT | S. aureus | 3.0 | 34 | 10:2 | 37 | 3 | 25-30 |
| NheI | G/CTAGC | M. bovis | 2.9 | 45 | 5:1 | 37 | 3 | 20-25 |
| NotI | GC/GGCCGC | M. bovis | 2.9 | 45 | 5:1 | 37 | 3 | 7 |
| SalI | G/TCGAC | M. bovis | 2.9 | 45 | 16:1 | 37 | 4 | » 15 |
| SmaI | CCC/GGG | S. aureus | 3.0 | 34 | 20:2 | 22 | 3 | 18 |
| SpeI | A/CTAGT | M. bovis | 2.9 | 45 | 5:1 | 37 | 3 | 20-25 |
| XbaI | T/CTAGA | M. bovis | 2.9 | 45 | 10:1 | 37 | 3 | 25-30 |
| XhoI | C/TCGAG | M. bovis | 2.9 | 45 | 16:1 | 37 | 4 | 15-20 |
*Perform 50°C digestions under mineral oil.
D. Genome Complexity and Expected Restriction Site Frequency
It is possible to calculate the expected average fragment size for a given genomic DNA if the percent GC content of the DNA and the recognition sequence of the restriction enzyme are known. For example, in a genome with 50% GC content and no dinucleotide bias, a four-cutter can be expected to cut every 44 bases (256), a six-cutter can be expected to digest every 46 (4,096) bases, and an eight-cutter should digest every 48 (65,536) bases. For sequences with GC contents other than 50% it is still possible to do this calculation by considering the probability of a particular nucleotide appearing at each position in the recognition sequence.
The general form of the equation is:
Expected cutting frequency = (0.5 x GC)a x (0.5 x AT)b
"GC" & "AT" are the probability that a given base is (G or C) or (A or T) (the GC or AT content of the target DNA), "a" is the number of G's and C's and "b" is the number of A's and T's in the restriction enzyme's recognition sequence.
For example, for an EcoRI digest (GAATTC) of DNA from an organism with 40% GC and no dinucleotide bias the expected chance of cutting would be:
(0.5 x GC)a x (0.5 x AT)b = (0.5 x 0.4)2 x (0.5 x 0.6)4 = 0.000324
The probability of cutting any given 6 base sequence is 0.000324, or an average of one cut every 3086 bases.
The equation can be refined if there is a known bias in the frequency of dinucleotide and trinucleotide repeats in the DNA being digested (5). For a sequence N1N2N3N4N5N6 (where N1 through N6 are the bases in the restriction enzyme recognition sequence), the expected frequency of digestion can be calculated as
p(N1N2) p(N2N3) p(N3N4) p(N4N5) p(N5N6)/p(N3) p(N4) p(N5)
Where p(N) is the frequency of N in the genome and p(NaNb) is the dinucleotide repeat frequency.
or
p(N1N2N3) p(N2N3N4) p(N3N4N5) p(N4N5N6)/p(N2N3) p(N3N4) p(N4N5)
Where p(NaNb) is the dinucleotide repeat frequency and p(NaNbNc) is the trinucleotide repeat frequency.
The GC content and dinucleotide frequencies of many organisms have been determined (6). Because the sequences of many organisms have been elucidated it is now possible to generate complete restriction maps of entire genomes. Most of the completed genome sequences are available from the World Wide Web from sites such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) at: http://www.genome.ad.jp/kegg/java/org_list.html
Other useful web sites include:
E. Analysis of Large DNA Fragments
Standard agarose gel electrophoresis can be used to resolve DNA in the ~10bp (4% gel) to ~50kb (0.3% gel) range. For resolution of larger DNA fragments it is necessary to use pulsed field gel electrophoresis (PFGE). PFGE relies on the observation that the rate of re-orientation of DNA within an electric field is proportional to the size of the DNA fragment. In PFGE, the orientation of the electric field relative to the gel, and thus the DNA, is changed throughout the gel run. Larger DNAs re-orient more slowly and thus have slower net migration rates. Several different types of PFGE including: orthogonal field agarose gel electrophoresis (OFAGE), field inversion gel electrophoresis (FIGE), rotating agarose gel electrophoresis (RAGE), and contour clamped homogenous electric field (CHEF) can be used for this purpose.
References
- Ausubel, F.M. et al. (1993) Current Protocols in Molecular Biology , Vol. 1, Greene Publishing Associates, Inc., and John Wiley and Sons, NY. 221, 317.
- Wizard® Genomic DNA Purification Kit Technical Manual, TM050, Promega Corporation.
- Protocols and Applications Guide, online edition. Promega Corporation.
- Anand, R. and Southern, E. (1990) In Gel Electrophoresis of Nucleic Acids: A Practical Approach, Second Edition. Rickwood, D., and Hames, B. eds. IRL Press, Oxford, U.K.
- Perbal, B. (1998) A Practical Guide to Molecular Cloning, John Wiley and Sons, NY. 327.
- McClelland, M. et al. (1987) Restriction endonucleases for pulsed field mapping of bacterial genomes. Nucleic Acids Res. 15, 5985–6005.
Digestion of Supercoiled Plasmid DNA
Restriction enzyme units are usually defined using linear DNA substrates containing multiple recognition sites as these tend to give more reproducible results. Lambda and Adenovirus are the two substrates used most frequently because of their commercial availability and high quality. One unit of enzyme is the amount necessary to completely digest 1µg of such DNA in one hour under the appropriate buffer and temperature conditions.
Molecular biology applications frequently involve cutting a supercoiled plasmid at a single site within the multiple cloning sequence. Often, more than 1 unit of enzyme is required to digest 1µg of plasmid. There are several reasons why this is the case. For example, there are 0.0317 picomoles of DNA in 1µg of lambda. HindIII cleaves this substrate 7 times or 0.2219 picomoles of recognition sites in 1µg. For a 3,000 base pair plasmid with a single recognition site, there are 0.5 picomoles of DNA in 1µg and also 0.5 picomoles of recognition sites, over twice as many as for the same mass of lambda DNA. The ability of a restriction enzyme to find a single site by linear diffusion in the supercoiled plasmid is also presumed to be different than for any of the sites on a linear substrate. Although it is not common, some enzymes exhibit differences in their ability to cut supercoiled DNA depending on the buffer conditions used. For example, SacII exhibits a pronounced difference in its ability to cut supercoiled plasmids depending on buffer conditions, but this sensitivity is not seen nearly as dramatically with linear substrates. Promega's Reaction Buffer C, supplied with SacII works well for both linear and supercoiled DNA substrates.
The Table below lists the minimum number of units necessary to completely cut 1µg of a supercoiled pGEM® Vector containing a single recognition site. Commonly used Promega restriction enzymes, including all those that are blue/white cloning qualified, are listed.
Minimum Number of Units of Enzyme Necessary to Cut 1µg of Supercoiled DNA Containing a Single Restriction Site |
|||
| Enzyme | Minimum Units |
Enzyme | Minimum Units |
| ApaI | 1 | NdeI |
4 |
| BamHI | 2 | NotI |
2 |
| BglII | 2 | PstI | 1 |
| ClaI | 1 | SacI | 4 |
| EcoRI | 2 | SacII | 20 |
| EcoRV | 1 | SalI | 5 |
| HindIII | 1 | SmaI | 1 |
| KpnI | 1 | SpeI | 1 |
| MluI | 1 | SphI | 1 |
| NcoI | 1 | XbaI | 1 |
| XhoI | 2 | ||
Digestion of Restriction Sites Close to the End of Linear DNA
In order to recognize and cleave their recognition sequence, most restriction enzymes need some flanking DNA. Because of this it can be difficult to achieve complete digestion of PCR products that have restriction sites engineered near the end of a primer or to perform double digests using two enzymes that cut at sites close to each other in a polylinker region. Such digestions may be improved by using long (16-hour) incubation times.
A. Multiple Digests
When performing multiple digests within a polylinker region, it is important to determine if the sites overlap such that cleavage at one site will destroy another. For example, the sequence below contains both a KpnI (GGTAC/C) and a SmaI (CCC/GGG) site.
...NNNNNGGTACCCGGGNNNNN...
...NNNNNCCATGGGCCCNNNNN...
If this DNA is first digested with KpnI, it will leave the following sequence, which cannot be digested with SmaI.
...NNNNNGGTAC CCGGGNNNNN...
...NNNNNC CATGGGCCCNNNNN...
Alternatively, if the DNA is first digested with SmaI, it will leave the sequence shown below, which can be digested with KpnI, although there may be problems due to a lack of flanking bases.
...NNNNNGGTACCC GGGNNNNN...
...NNNNNCCATGGG CCCNNNNN...
Studies by Kaufman and Evans (1), and Moreira and Noren (2) show the efficiency of digestion of polylinker regions with a variety of enzymes. This data can be used to help determine the order in which two enzymes should be used for the most efficient multiple digests, or to predict whether enzymes will work effectively in a double-digest. Care must be taken when applying the conclusions from these publications to the digestion of PCR products because the majority of the ends left by restriction enzymes have 2-4 base 3´ or 5´ overhangs. Generally, PCR products are either blunt ended (if a proofreading thermostable polymerase is used) or contain a single 3´ overhanging base (if a non-proofreading enzyme is used).
B. PCR Products
In general, the addition of 2-6 extra bases upstream of an engineered restriction site in a PCR primer will greatly increase the efficiency of digestion of the amplification product, but this is dependent on the enzyme used. Table 2.6 shows the results of a study where the ability of restriction enzymes to digest various PCR products was tested (3). PCR products in which the first base pair of the restriction site was flush with (0), or 1, 2, or 3 base pairs away from the end of the fragment were tested with a variety of enzymes.
Ability of Restriction Enzymes to Cut PCR Products that have Engineered Restriction Sites Near the End of the Fragment |
||||
| Enzyme | Distance (in bp) from the End of the PCR Fragment | |||
| 0 | 1 | 2 | 3 | |
| ApaI | – | – | ± | + |
| BamHI | – | ± | + | + |
| ClaI | – | ± | + | + |
| EcoRI | – | ± | + | + |
| EcoRV | – | + | + | + |
| HindIII | – | – | + | + |
| NotI | – | – | + | + |
| PstI | – | – | ± | + |
| SacI | – | ± | + | + |
| SalI | + | + | + | + |
| SmaI | – | ± | + | + |
| SpeI | + | + | + | + |
| XbaI | – | ± | + | + |
| XhoI | – | – | ± | + |
Purified PCR fragments (10-50ng) were digested at least twice with 0.5 units of restriction enzyme in 10µl of the appropriate reaction buffer for 45 minutes. Digestion is indicated as follows: cleavable (+), not cleavable (–), and not reproducible (±). Table reproduced by permission of Eaton Publishing.
The addition of upstream bases to PCR primers is not the only method used to improve digestion efficiency. A number of protocols have been proposed to improve digestion including proteinase K treatment to remove any thermostable polymerase that may be blocking the DNA, end-polishing with Klenow or T4 DNA Polymerase and the addition of spermidine. However, none of these methods have been shown to improve cloning efficiency significantly (4) (5).
An additional drawback to the incorporation of restriction enzyme sites in PCR primers is that it can be quite difficult to resolve digested PCR products from those that remain uncut. This can be overcome by the addition of fluorescent tags at the 5´ ends of the primers prior to PCR. This allows identification of products that have been cut successfully because the label is lost upon digestion (6) .
An alternative method that has been used successfully to improve digestion of PCR products is to concatemerize the fragments after amplification (1) (5). This is achieved by first treating the cleaned up PCR products with T4 Polynucleotide Kinase (if the primers have not already been phosphorylated). The ends will already be blunt if a proofreading thermostable polymerase such as Pfu was used or may be treated with T4 DNA Polymerase to polish the ends if a non-proofreading polymerase such as Taq was used. PCR products are then ligated with T4 DNA ligase. This effectively moves the restriction enzyme sites away from the ends of the fragments and allows efficient digestion.
Back to topReferences
- Kaufman, D.L. and Evans, G.A. (1990) Restriction endonuclease cleavage at the termini of PCR products. BioTechniques 9, 304, 306.
- Moreira, R.F. and Noren, C.J. (1995) Minimum duplex requirements for restriction enzyme cleavage near the termini of linear DNA fragments. BioTechniques 19, 56, 58–9.
- Zimmermann, K. et al. (1998) Digestion of terminal restriction endonuclease recognition sites on PCR products. BioTechniques 24, 582–4.
- Jung, V. et al. (1990) Efficient cloning of PCR generated DNA containing terminal restriction endonuclease recognition sites. Nucleic Acids Res. 18, 6156.
- Jung, V. et al. (1993) Cloning of polymerase chain reaction-generated DNA containing terminal restriction endonuclease recognition sites. Meth. Enzymol. 218, 357–62.
- Yamaguchi, K. et al. (1994) Fluorescent primers allow direct confirmation of restriction enzyme cleavage of PCR products. BioTechniques 17, 640–50, 652.
Troubleshooting Restriction Enzyme Digestions
This troubleshooting guide addresses common problems that may be encountered while using restriction enzymes. If problems persist contact Promega Technical Services at techserv@promega.com or 800-356-9526 (U.S. and Canada only).
| Problem: No cleavage | |
| Probable Cause | Comments |
| Dirty template DNA | Clean up the substrate DNA using the Wizard® DNA Clean-Up System (Cat.# A7280). Alternatively, phenol/chloroform extraction followed by ethanol precipitation can be used to purify the DNA substrate. A discussion of substrate quality and considerations can be found in the Substrate Considerations section of this guide. |
| Presence of inhibitors | Enzyme inhibitors in the substrate DNA solution (e.g., SDS, phenol, EDTA, chloroform, ethanol, CsCl, high salt, or plasticizers from microcentrifuge tubes) can be removed using the Wizard® DNA Clean-Up System. Alternatively, phenol/chloroform extraction and/or ethanol precipitation can be used to remove inhibitors from the DNA preparation. |
| DNA methylated (e.g. dam, dcm) | Check the Methylation Sensitivity Reference Table for information on the sensitivity of the restriction enzyme to the methylation state of the substrate. For plasmid DNA, eliminate methylation by passaging the DNA through a bacterial host that is deficient in the interfering methylase. For example, E. coli JM110, which lacks both dam and dcm activity. Digest the DNA using an isoschizomer that is insensitive to methylation. Consult the Isoschizomers Reference Table for isoschizomer enzyme pairs that differ in their ability to cut methylated DNA. |
| DNA unmethylated | Some enzymes require methylation of their target sites. For example, Dpn I requires N6-methylation of the adenine residue for activity. See the Methylation Sensitivity Reference Table for further information on the effect of site-specific methylation on Promega restriction enzymes. |
| Inactive enzyme | Test the enzyme on substrate DNA that has been digested successfully in the past or test the enzyme with the DNA substrate used for determination of the enzyme unit activity. Usually this is Lambda DNA (Cat.# D1501). |
| Suboptimal reaction conditions | Consult the Promega Product Information sheet provided with the enzyme for recommended reaction conditions. Suggested reaction conditions can be found in the Standard Reactions section of this guide. |
| Incorrect sequence information | Double check sequence information to confirm the number and location of enzyme recognition sites. |
| Problem: Partial Cleavage. | |
| Probable Cause | Comments |
| Dirty template DNA | Clean up the substrate DNA using the Wizard® DNA Clean-Up System (Cat.# A7280). Alternatively, phenol/chloroform extraction followed by ethanol precipitation can be used to purify the DNA substrate. A discussion of substrate quality and considerations can be found in the Substrate Considerations section of this guide. |
| Loss of restriction enzyme activity | Digest substrate DNA with several other restriction enzymes to ensure that impurities in the digest are not interfering with enzyme activity. Alternatively, the enzyme activity can be tested using the unit activity assay conditions. The Promega Product Information sheet supplied with the enzyme contains information on the unit activity assay conditions specific for that enzyme. See the comments under the problem Enzyme activity lower than expected for more possible causes and solutions for low restriction enzyme activity. |
| Presence of enzyme inhibitors | Enzyme inhibitors in the substrate DNA (e.g., SDS, phenol, EDTA, chloroform, ethanol, CsCl, high salt, or plasticizers from microcentrifuge tubes) can be removed using the Wizard® DNA Clean-Up System (Cat.# A7280). Alternatively, phenol/chloroform extraction and/or ethanol precipitation can be used to remove inhibitors from the DNA preparation. |
| Improper reaction conditions | Check that proper reaction conditions were used including the optimal buffer, temperature and amount of enzyme. Suggested reaction conditions can be found in the Standard Reactions section of this guide or on the Promega Product Information sheet provided with each restriction enzyme. |
| Restriction enzyme not completely mixed into reaction | Add the restriction enzyme to the digest last and mix gently. Ensure that the enzyme is mixed thoroughly into the reaction but do not vortex. |
| Loss of restriction enzyme activity upon dilution prior to use | If possible, do not dilute the enzyme prior to use. If the enzyme must be diluted, use the recommended storage buffer for that enzyme. If used immediately, enzyme can be diluted in Reaction Buffer containing 0.5mg/ml Acetylated BSA. Enzymes diluted into the reaction buffer do not store well. Never dilute the enzyme directly in water. Mix gently, do not vortex. |
| Loss of restriction enzyme activity upon addition to digest | Enzyme has lost activity upon dilution into reaction. Use optimum restriction enzyme buffer supplemented with 0.1mg/ml Acetylated BSA to stabilize enzyme in the reaction. |
| DNA concentration too high | Reduce DNA concentration or use multiple reactions. Viscous DNA solutions can inhibit enzyme digestions. |
| DNA concentration too low | Sample DNA concentration is below the Km of the restriction enzyme. Add more DNA to the reaction. |
| Annealed DNA ends (e.g., lambda DNA) | The ends of some DNA substrates such as the cos ends of lambda may re-anneal during digestion. This can give the appearance that digestion is incomplete. Heat the DNA at 65°C for 5 minutes prior to gel electrophoresis to melt ends that have annealed. The presence of restriction enzyme buffer is important while heating as this will prevent small DNA fragments from melting into single-strands. |
| Denaturation of restriction enzyme | Many restriction enzymes can be inactivated by heat. Also, avoid vortexing dilutions or reactions containing restriction enzymes. |
| DNA substrate is supercoiled | Supercoiled DNA generally requires more units of enzyme than linear DNA for complete digestion. The unit activity of restriction enzymes is determined using linear DNA templates. One unit of restriction enzyme may cut one microgram of linear DNA in one hour, but this may not be true of supercoiled DNA. See Digestion of Supercoiled Plasmid DNA for further information. Alternatively, try using five units of restriction enzyme per microgram of supercoiled DNA for digestion in one hour. Another option is to linearize the DNA with an enzyme that is not resistant to supercoiling, then digest with the enzyme of choice. Alternatively, relax the DNA with topoisomerase, then digest with the restriction enzyme. |
| Substrate DNA has many restriction sites per unit of mass | The Promega Product Information sheet supplied with each enzyme lists the substrate DNA used in the unit activity assay and how many cut sites the substrate has for that enzyme. While one unit of enzyme will cut 1µg of the activity assay substrate to completion in one hour, DNA with many more cut sites per microgram will require more units of enzyme for complete digestion in one hour. See Substrate Considerations for further information. The optimum amount of enzyme should be determined for each substrate. |
| Problem: Enzyme activity lower than expected. | |
| Probable Cause | Comments |
| Suboptimal reaction conditions | Consult the Promega Product Information sheet provided with the enzyme for recommended reaction conditions. Suggested reaction conditions also can be found in the Standard Reactions section of this guide. |
| Incorrect storage or handling of enzyme | Store all restriction enzymes at -20°C in a non-frost free freezer. Remove the enzyme just prior to use and keep on ice. Return the enzyme to the freezer as soon as possible. Do not vortex the enzyme or the reaction mix containing the enzyme. Instead, mix by gentle pipetting. Avoid air bubbles. |
| Enzyme stored diluted | It is best to store the enzymes as supplied in concentrated form. If the enzyme must be diluted, use the recommended enzyme storage buffer. If used immediately, enzyme can be diluted in Reaction Buffer containing 0.5mg/ml BSA. Enzymes diluted in Reaction Buffer do not store well. |
| Incorrect dilution of enzyme | If enzyme is diluted just prior to use, dilute the enzyme in the recommended 1X Reaction Buffer supplemented with 0.5mg/ml Acetylated BSA. Never dilute enzyme directly in water. Check the dilution factor used to ensure that the enzyme concentration is correct. Use the enzyme as soon as possible after dilution. |
| Pipetting error | Use a positive displacement pipet for viscous solutions such as concentrated DNA and enzyme storage buffer, which contains 50% glycerol. |
| Glycerol inhibition | Inhibition of the reaction may occur if the volume of enzyme added is greater than 10% of the total reaction volume. |
| Reaction temperature suboptimal | Check for optimal reaction temperature on the Promega Product Information sheet supplied with the enzyme. See Relative Activity of Restriction Enzymes in Promega 10X Buffers Reference Table for a listing of optimal temperatures for Promega restriction enzymes. |
| DNA substrate is supercoiled | Supercoiled DNA generally requires more units of enzyme than linear DNA for complete digestion. The unit activity of restriction enzymes is determined using linear DNA substrates. One unit of restriction enzyme may cut one microgram of linear DNA in one hour, but this may not be true of supercoiled DNA. See Digestion of Supercoiled Plasmid DNA for further information. Alternatively, try using five units of restriction enzyme per microgram of supercoiled DNA for digestion in one hour. |
| Substrate DNA has many restriction sites per unit of mass | The Promega Product Information sheet supplied with each enzyme lists the substrate DNA used in the unit activity assay and how many cut sites the substrate has for that enzyme. While one unit of enzyme will cut 1µg of the activity assay substrate to completion in one hour, DNA with many more cut sites per microgram will require more units of enzyme for complete digestion in the same time. See Substrate Considerations for further information. The optimum amount of enzyme should be determined for each substrate. Guidelines for Digestion of High Molecular Weight DNA can be found elsewhere in this guide. |
| Suboptimal reaction conditions | Consult the Promega Product Information sheet provided with the enzyme for recommended reaction conditions. Suggested reaction conditions can be found in the Standard Reactions section of this guide. |
| Problem: Greater than expected number of DNA fragments. | |
| Probable Cause | Comments |
| Star activity | Star activity or relaxed specificity of the restriction enzyme for its cognate sequence is caused by suboptimal digestion conditions. Common causes of star activity include the use of excess enzyme (generally >100units/µg), excess glycerol (>5% v/v), the presence of manganese or other divalent cation instead of magnesium, or nonoptimal NaCl concentrations. Extremes of pH (especially >pH 8.0) and the presence of DMSO, ethanol or other organic solvents are also causes of star activity. For more information, see Star Activity. To avoid star activity, use the recommended digestion conditions for the enzyme and avoid using DNA substrates that may be contaminated with salts or solvents. Conditions for setting up a restriction enzyme digest can be found in the Standard Reactions section of this guide and on the Promega Product Information sheet supplied with each enzyme. |
| Sample contaminated with another DNA | Confirm the activity of the restriction enzyme by testing either the substrate used for the unit activity assay (generally lambda DNA) or another substrate known to contain a single DNA species. If the digestion pattern is correct for these substrates then the extra bands present may be due to another DNA contaminating the reaction. Test the enzyme, reaction buffers and gel loading buffer for DNA contamination. |
| Presence of a second restriction enzyme | Detect a second activity by repeating the unit activity assay or test a known DNA substrate with a defined number of cut sites for the enzyme. |
| Volume of reaction decreased during long digestion | During extended digestions the volume of the digest may decrease, especially if a thermophilic restriction enzyme is used. A reduction in the volume of the reaction may lead to star activity by concentrating the glycerol, enzyme, salt, or any contaminants in the reaction. For long digestions or digestions at elevated temperatures, add mineral oil to the surface, decrease the incubation time of the reaction or perform the reaction in an incubator. |
| Incorrect sequence information | Double check sequence information to confirm the number and location of enzyme recognition sites. |
| Problem: No DNA observed after digestion. | |
| Probable Cause | Comments |
| Concentration of DNA substrate incorrect | Determine the concentration of the DNA used in the digestion by spectrophotometry or gel electrophoresis prior to digestion. Excessive RNA or salt (e.g., guanidine) contamination of the DNA sample will increase the absorbance at 260nm, leading to an artificially high determination of concentration. Confirm DNA concentration by electrophoresis. If necessary, treat the sample with RNase or ethanol-precipitate to remove RNA or salts, respectively. |
| Nuclease contamination from bacterial host | If the DNA was isolated from an endA(+) bacterial strain, it may be contaminated with endonuclease I. If so, when magnesium is present (as in a restriction enzyme digest), the endonuclease will be activated and the DNA substrate digested. If possible use an endA(-) strain for propagation of the DNA substrate. The Wizard® Plus DNA purification products can be used with endA(+) strains to produce endonuclease-free DNA. Consult the Wizard® Plus SV Minipreps DNA Purification System Technical Bulletin, #TB225 for more information. Alternatively phenol/chloroform extract the DNA sample before digestion to eliminate endonuclease I contamination. |
| Nuclease contamination from reagents | Test individual reaction components for nuclease contamination. Bacterial and fungal contamination is often the source of nucleases. |
| Problem: A smear of DNA is observed after digestion. | |
| Probable Cause | Comments |
| Nuclease contamination from bacterial host or nuclease contamination from reagents | Complete nonspecific digestion will result in disappearance of the DNA substrate. Partial digestion will result in a smear of DNA being observed from a point at the estimated size of the DNA substrate to the bottom of the gel. |